TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, Maryland 21218, United States.
J Phys Chem B. 2021 Apr 22;125(15):3739-3751. doi: 10.1021/acs.jpcb.0c10992. Epub 2021 Apr 7.
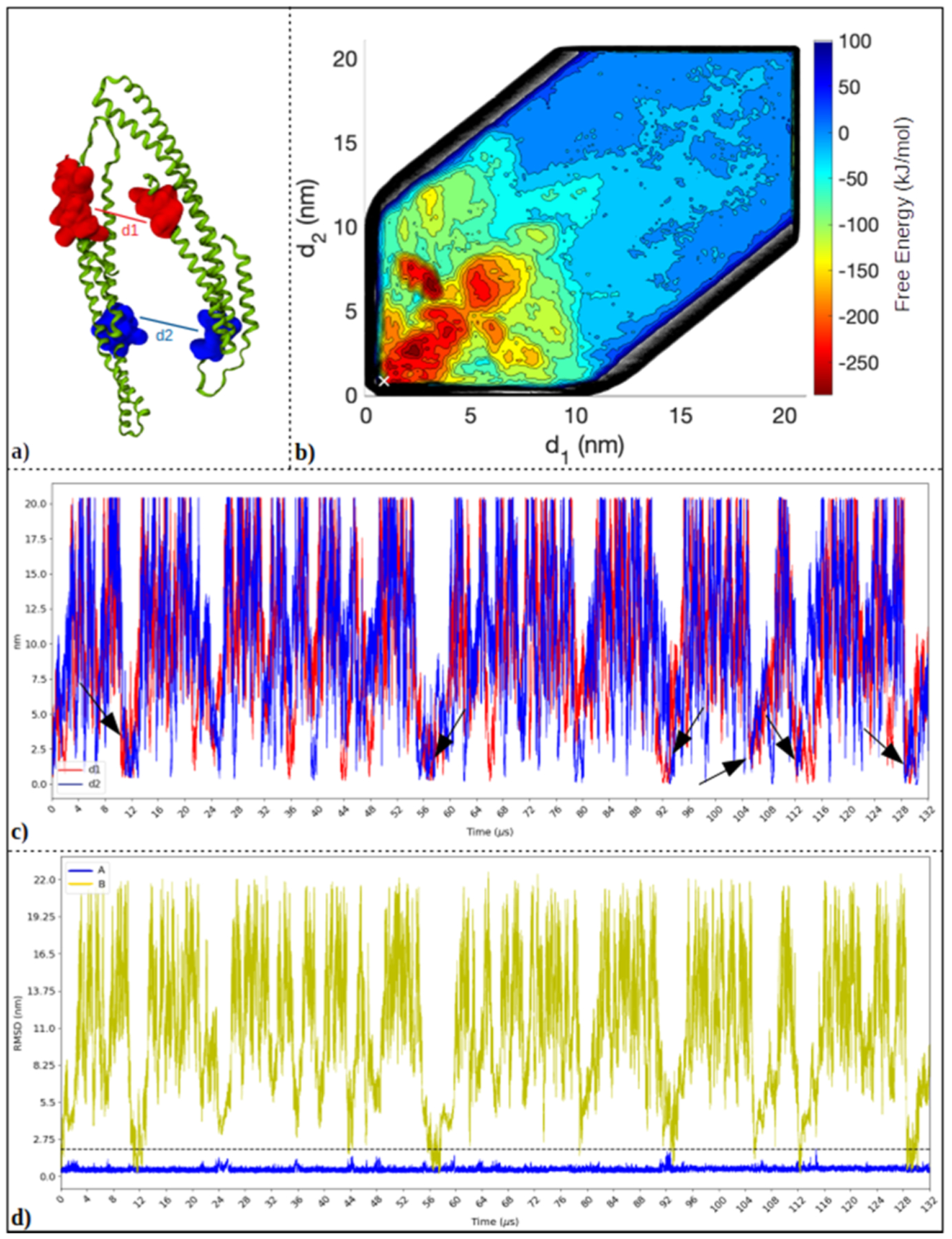
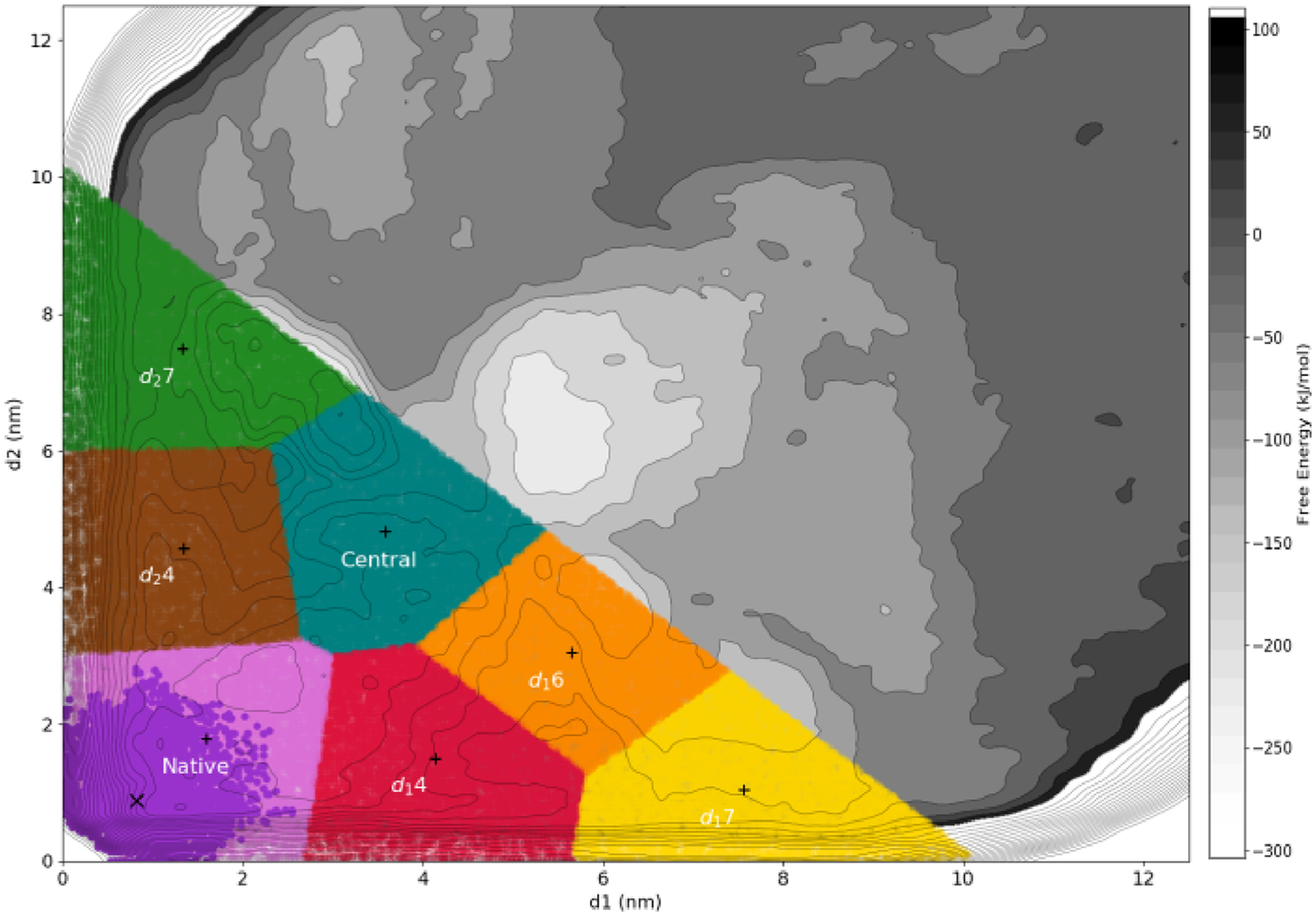
Proteins with BAR domains function to bind to and remodel biological membranes, where the dimerization of BAR domains is a key step in this function. These domains can dimerize in solution or after localizing to the membrane surface. Here, we characterize the binding thermodynamics of homodimerization between the LSP1 BAR domain proteins in solution, using molecular dynamics (MD) simulations. By combining the MARTINI coarse-grained protein models with enhanced sampling through metadynamics, we construct a two-dimensional free energy surface quantifying the bound versus unbound ensembles as a function of two distance variables. With this methodology, our simulations can simultaneously characterize the structures and relative stabilities of a range of sampled dimers, portraying a heterogeneous and extraordinarily stable bound ensemble, where the proper crystal structure dimer is the most stable in a 100 mM NaCl solution. Nonspecific dimers that are sampled involve contacts that are consistent with experimental structures of higher-order oligomers formed by the LSP1 BAR domain. Because the BAR dimers and oligomers can assemble on membranes, we characterize the relative alignment of the known membrane binding patches, finding that only the specific dimer is aligned to form strong interactions with the membrane. Hence, we would predict a strong selection of the specific dimer in binding to or assembling when on the membrane. Establishing the pairwise stabilities of homodimer contacts is difficult experimentally when the proteins form stable oligomers, but through the method used here, we can isolate these contacts, providing a foundation to study the same interactions on the membrane.
具有 BAR 结构域的蛋白质能够结合和重塑生物膜,而 BAR 结构域的二聚化是其功能的关键步骤。这些结构域可以在溶液中或在定位于膜表面后二聚化。在这里,我们使用分子动力学 (MD) 模拟来描述 LSP1 BAR 结构域蛋白在溶液中的同源二聚化的结合热力学。通过将 MARTINI 粗粒化蛋白质模型与通过元动力学增强的采样相结合,我们构建了一个二维自由能表面,该表面将结合态与非结合态的集合作为两个距离变量的函数进行量化。通过这种方法,我们的模拟可以同时描绘出一系列所采样的二聚体的结构和相对稳定性,描绘出一个异质且非常稳定的结合态集合,其中在 100mM NaCl 溶液中,适当的晶体结构二聚体是最稳定的。所采样的非特异性二聚体涉及与由 LSP1 BAR 结构域形成的高阶寡聚物的实验结构一致的接触。由于 BAR 二聚体和寡聚体可以在膜上组装,我们对已知的膜结合斑块的相对取向进行了特征描述,发现只有特定的二聚体与膜对齐以形成强相互作用。因此,我们可以预测在结合或组装到膜上时,特异性二聚体的强烈选择。当蛋白质形成稳定的寡聚体时,通过实验确定同源二聚体接触的成对稳定性是困难的,但通过这里使用的方法,我们可以分离这些接触,为在膜上研究相同的相互作用提供基础。