Blin Marion, Lacroix Laurent, Petryk Nataliya, Jaszczyszyn Yan, Chen Chun-Long, Hyrien Olivier, Le Tallec Benoît
Département de Gastro-entérologie, pôle MAD, Assistance Publique des Hôpitaux de Marseille, Centre Hospitalier Universitaire de Marseille, Marseille, France.
Institut de Biologie de l'Ecole Normale Supérieure (IBENS), Ecole Normale Supérieure, CNRS, INSERM, Université PSL, 46 rue d'Ulm, F-75005 Paris, France.
Nucleic Acids Res. 2021 Jul 9;49(12):e69. doi: 10.1093/nar/gkab219.
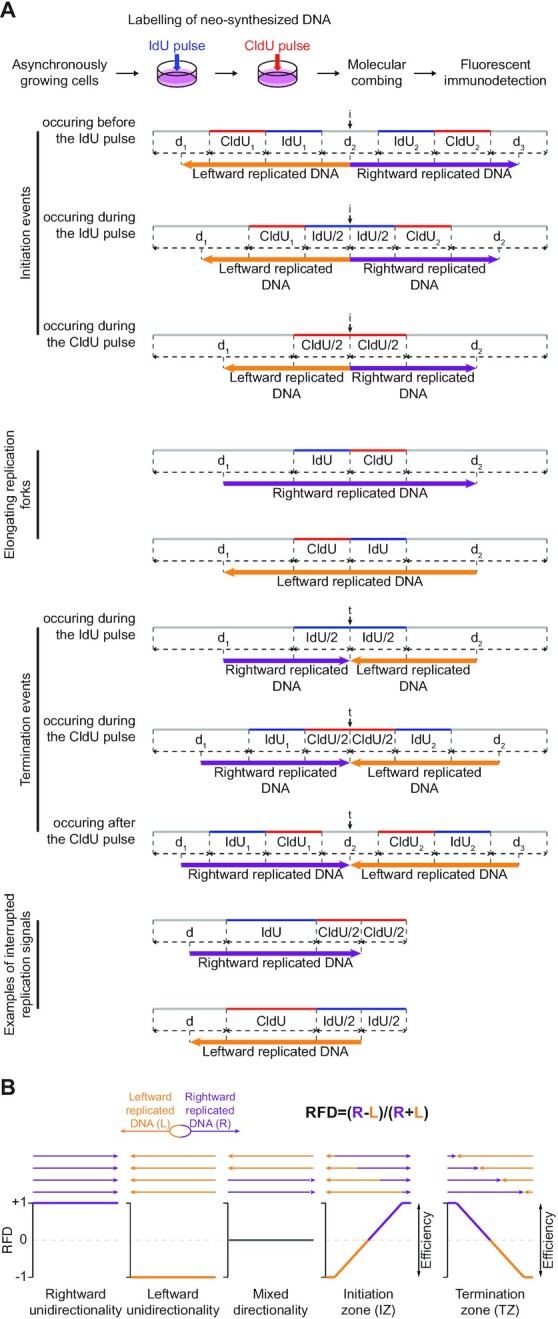
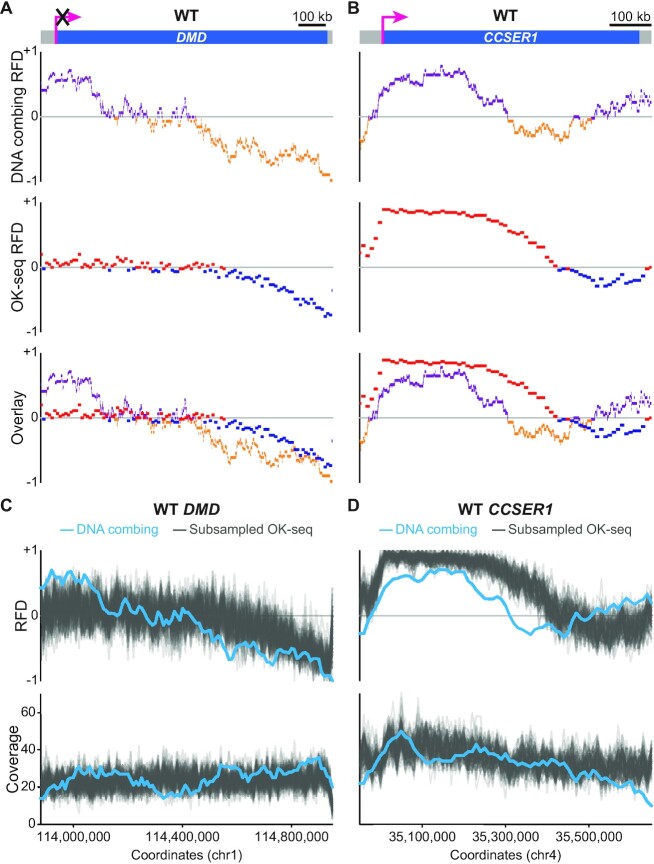
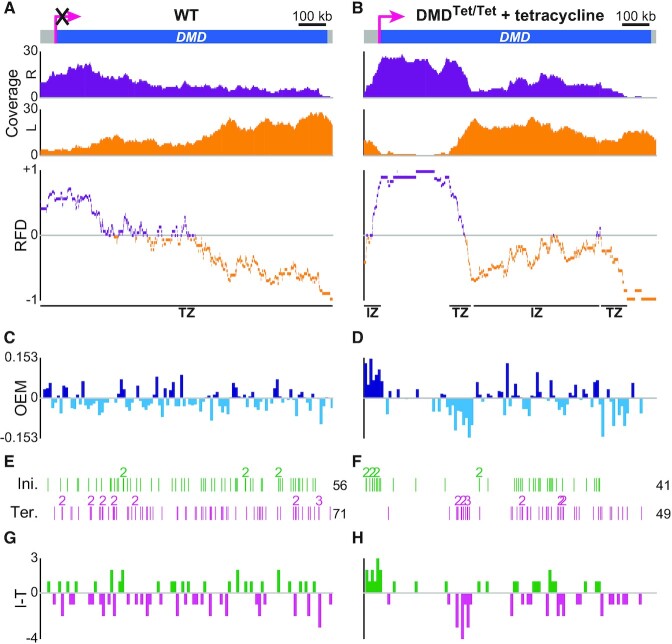
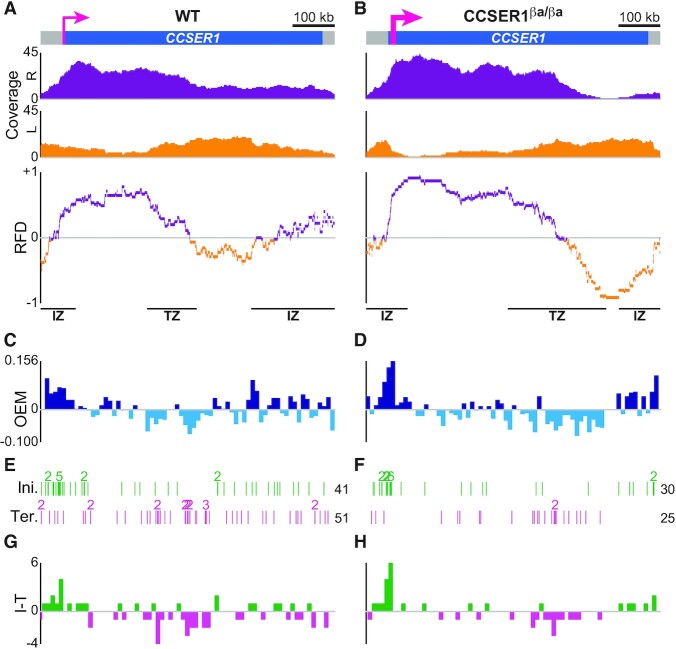
The replication strategy of metazoan genomes is still unclear, mainly because definitive maps of replication origins are missing. High-throughput methods are based on population average and thus may exclusively identify efficient initiation sites, whereas inefficient origins go undetected. Single-molecule analyses of specific loci can detect both common and rare initiation events along the targeted regions. However, these usually concentrate on positioning individual events, which only gives an overview of the replication dynamics. Here, we computed the replication fork directionality (RFD) profiles of two large genes in different transcriptional states in chicken DT40 cells, namely untranscribed and transcribed DMD and CCSER1 expressed at WT levels or overexpressed, by aggregating hundreds of oriented replication tracks detected on individual DNA fibres stretched by molecular combing. These profiles reconstituted RFD domains composed of zones of initiation flanking a zone of termination originally observed in mammalian genomes and were highly consistent with independent population-averaging profiles generated by Okazaki fragment sequencing. Importantly, we demonstrate that inefficient origins do not appear as detectable RFD shifts, explaining why dispersed initiation has remained invisible to population-based assays. Our method can both generate quantitative profiles and identify discrete events, thereby constituting a comprehensive approach to study metazoan genome replication.
后生动物基因组的复制策略仍不清楚,主要是因为缺少复制起点的确定性图谱。高通量方法基于群体平均值,因此可能只能识别高效的起始位点,而低效的起点则无法检测到。对特定基因座的单分子分析可以检测目标区域内常见和罕见的起始事件。然而,这些分析通常集中于定位单个事件,这只能给出复制动态的概述。在这里,我们通过汇总在分子梳拉伸的单个DNA纤维上检测到的数百条定向复制轨迹,计算了鸡DT40细胞中处于不同转录状态的两个大基因的复制叉方向性(RFD)图谱,即未转录和转录的DMD以及以野生型水平表达或过表达的CCSER1。这些图谱重建了由起始区域侧翼的终止区域组成的RFD结构域,该结构域最初在哺乳动物基因组中观察到,并且与由冈崎片段测序产生的独立群体平均图谱高度一致。重要的是,我们证明低效的起点不会表现为可检测到的RFD变化,这解释了为什么分散起始在基于群体的检测中仍然不可见。我们的方法既可以生成定量图谱,又可以识别离散事件,从而构成了一种研究后生动物基因组复制的综合方法。