Arora Uma P, Charlebois Caleigh, Lawal Raman Akinyanju, Dumont Beth L
The Jackson Laboratory, 600 Main Street, Bar Harbor, ME, 04609, USA.
Tufts University, Graduate School of Biomedical Sciences, 136 Harrison Ave, Boston, MA, 02111, USA.
BMC Genomics. 2021 Apr 17;22(1):279. doi: 10.1186/s12864-021-07591-5.
Mammalian centromeres are satellite-rich chromatin domains that execute conserved roles in kinetochore assembly and chromosome segregation. Centromere satellites evolve rapidly between species, but little is known about population-level diversity across these loci.
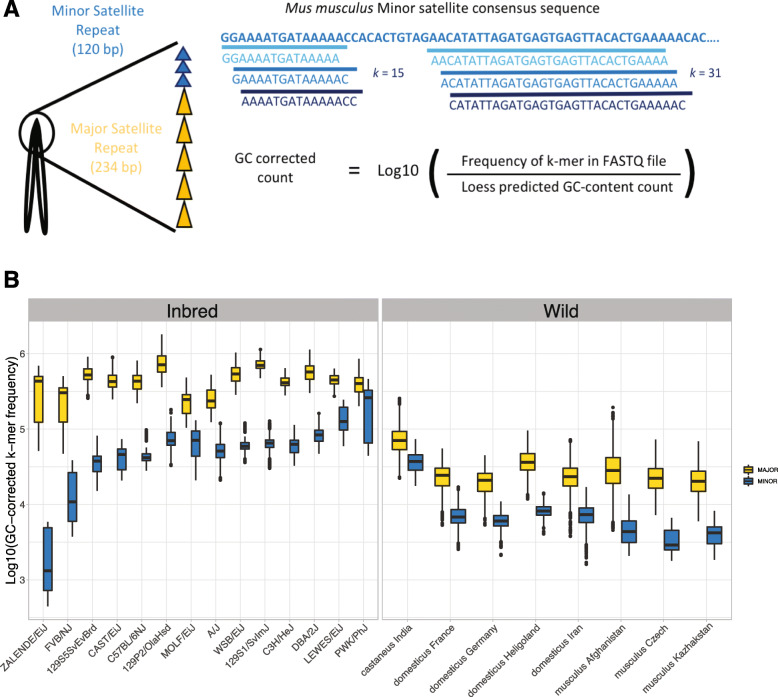
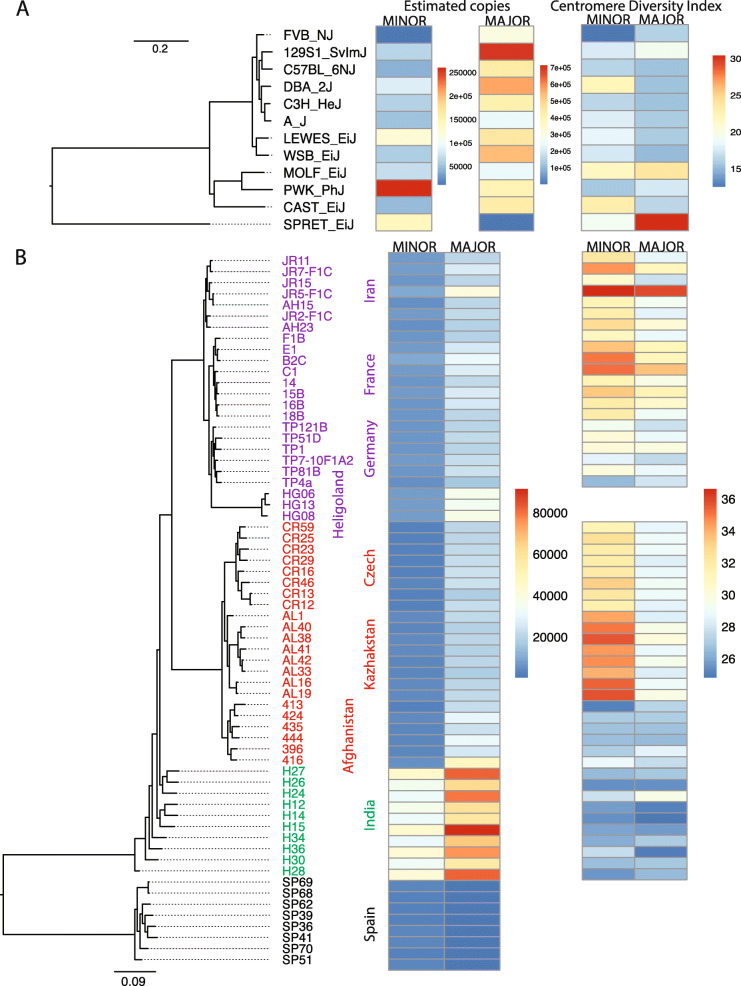
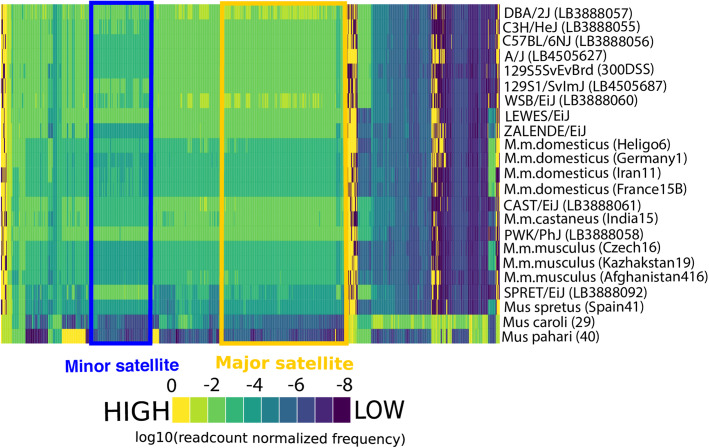
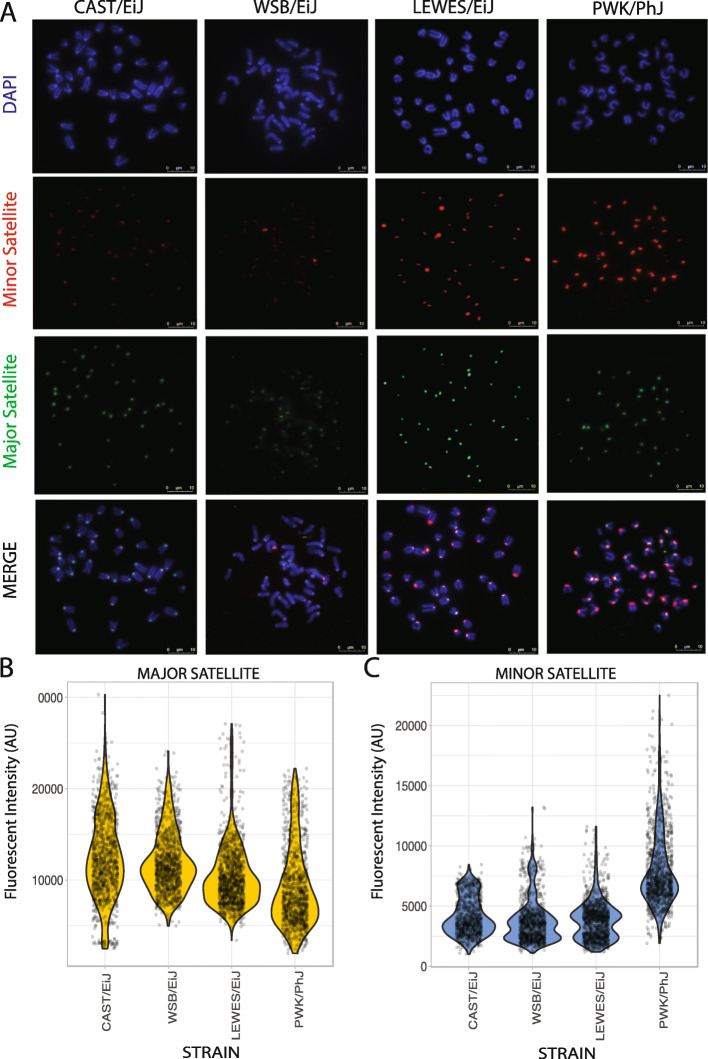
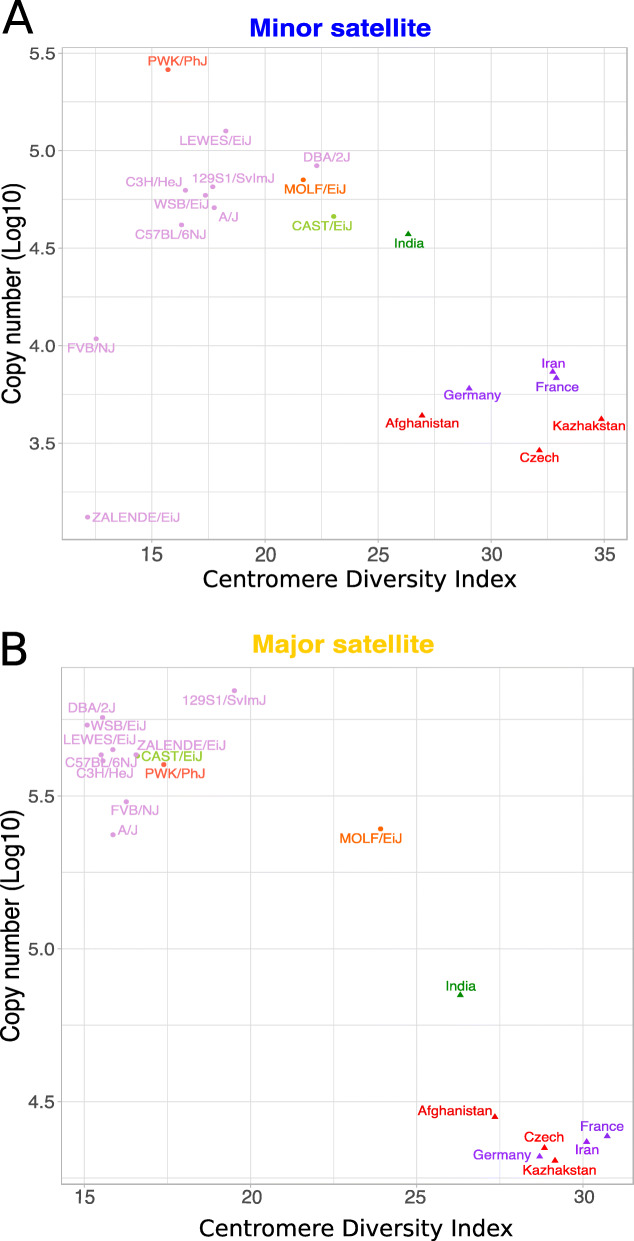
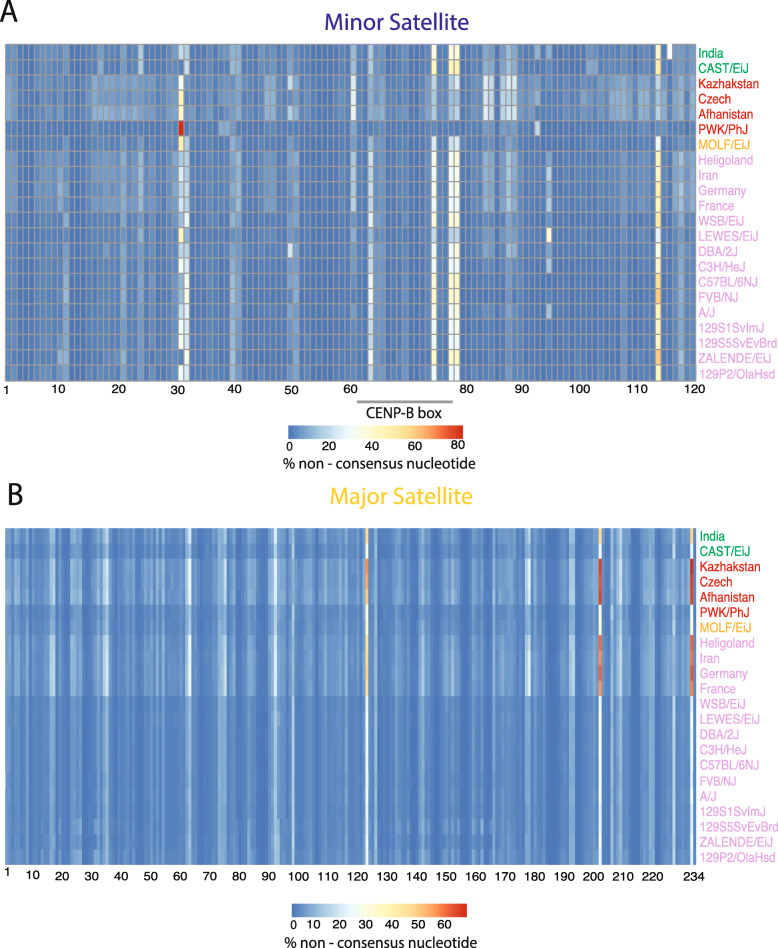
We developed a k-mer based method to quantify centromere copy number and sequence variation from whole genome sequencing data. We applied this method to diverse inbred and wild house mouse (Mus musculus) genomes to profile diversity across the core centromere (minor) satellite and the pericentromeric (major) satellite repeat. We show that minor satellite copy number varies more than 10-fold among inbred mouse strains, whereas major satellite copy numbers span a 3-fold range. In contrast to widely held assumptions about the homogeneity of mouse centromere repeats, we uncover marked satellite sequence heterogeneity within single genomes, with diversity levels across the minor satellite exceeding those at the major satellite. Analyses in wild-caught mice implicate subspecies and population origin as significant determinants of variation in satellite copy number and satellite heterogeneity. Intriguingly, we also find that wild-caught mice harbor dramatically reduced minor satellite copy number and elevated satellite sequence heterogeneity compared to inbred strains, suggesting that inbreeding may reshape centromere architecture in pronounced ways.
Taken together, our results highlight the power of k-mer based approaches for probing variation across repetitive regions, provide an initial portrait of centromere variation across Mus musculus, and lay the groundwork for future functional studies on the consequences of natural genetic variation at these essential chromatin domains.
哺乳动物的着丝粒是富含卫星序列的染色质结构域,在动粒组装和染色体分离中发挥着保守作用。着丝粒卫星序列在物种间进化迅速,但对于这些位点在种群水平上的多样性了解甚少。
我们开发了一种基于k-mer的方法,用于从全基因组测序数据中量化着丝粒拷贝数和序列变异。我们将此方法应用于不同的近交系和野生家鼠(小家鼠)基因组,以描绘核心着丝粒(次要)卫星序列和着丝粒周围(主要)卫星重复序列的多样性。我们发现,近交系小鼠品系中次要卫星序列的拷贝数变化超过10倍,而主要卫星序列的拷贝数范围为3倍。与关于小鼠着丝粒重复序列同质性的普遍假设相反,我们在单个基因组中发现了明显的卫星序列异质性,次要卫星序列的多样性水平超过了主要卫星序列。对野生捕获小鼠的分析表明,亚种和种群来源是卫星序列拷贝数变异和卫星序列异质性的重要决定因素。有趣的是,我们还发现,与近交系小鼠相比,野生捕获小鼠的次要卫星序列拷贝数显著减少,卫星序列异质性增加,这表明近亲繁殖可能以显著方式重塑着丝粒结构。
综上所述,我们的结果突出了基于k-mer方法在探测重复区域变异方面的强大功能,提供了小家鼠着丝粒变异的初步概况,并为未来研究这些重要染色质结构域自然遗传变异后果的功能研究奠定了基础。