Li Yue, Shen Ruoyi, Wang Anqi, Zhao Jian, Zhou Jieqi, Zhang Weijie, Zhang Ruochen, Zhu Jianjie, Liu Zeyi, Huang Jian-An
Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Soochow University, Suzhou, China.
Suzhou Key Laboratory for Respiratory Diseases, Suzhou, China.
Front Cell Dev Biol. 2021 Mar 18;9:648806. doi: 10.3389/fcell.2021.648806. eCollection 2021.
Lung adenocarcinoma (LUAD) originates mainly from the mucous epithelium and glandular epithelium of the bronchi. It is the most common pathologic subtype of non-small cell lung cancer (NSCLC). At present, there is still a lack of clear criteria to predict the efficacy of immunotherapy. The 5-year survival rate for LUAD patients remains low.
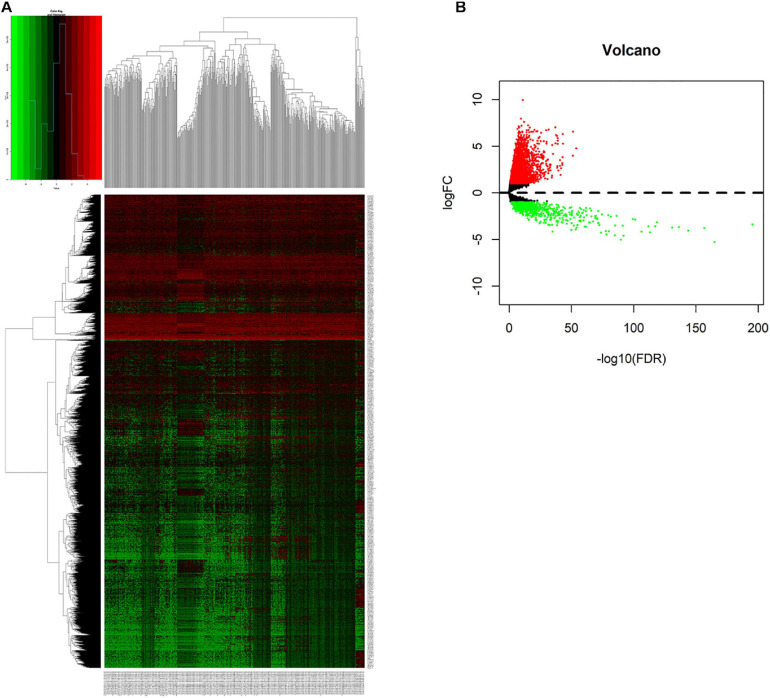
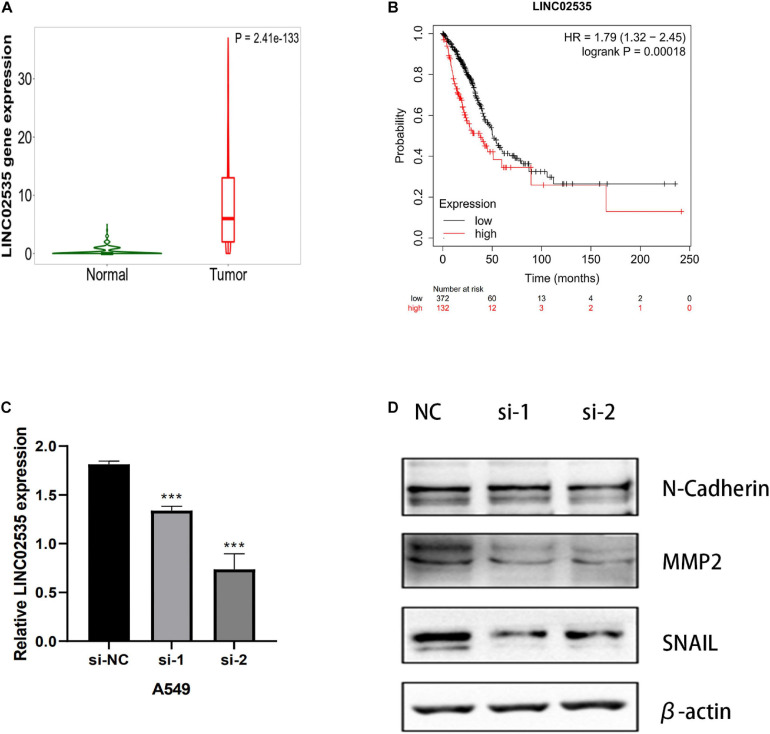
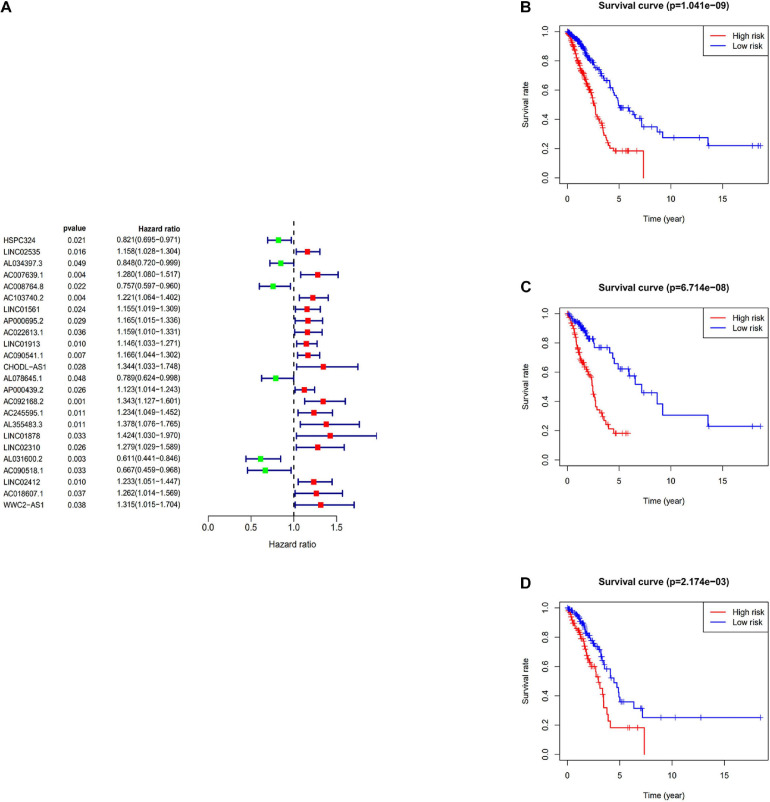
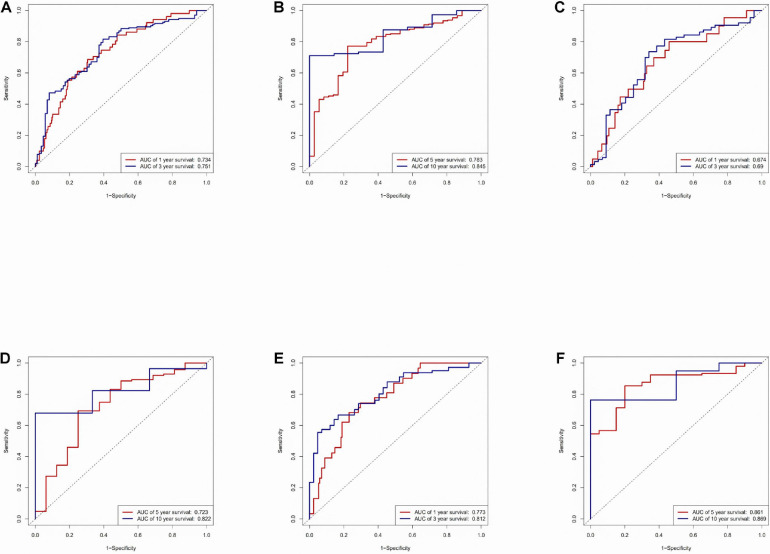
All data were downloaded from The Cancer Genome Atlas (TCGA) database. We used Gene Set Enrichment Analysis (GSEA) database to obtain immune-related mRNAs. Immune-related lncRNAs were acquired by using the correlation test of the immune-related genes with R version 3.6.3 (Pearson correlation coefficient cor = 0.5, < 0.05). The TCGA-LUAD dataset was divided into the testing set and the training set randomly. Based on the training set to perform univariate and multivariate Cox regression analyses, we screened prognostic immune-related lncRNAs and given a risk score to each sample. Samples were divided into the high-risk group and the low-risk group according to the median risk score. By the combination of Kaplan-Meier (KM) survival curve, the receiver operating characteristic (ROC) (AUC) curve, the independent risk factor analysis, and the clinical data of the samples, we assessed the accuracy of the risk model. Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed on the differentially expressed mRNAs between the high-risk group and the low-risk group. The differentially expressed genes related to immune response between two risk groups were analyzed to evaluate the role of the model in predicting the efficacy and effects of immunotherapy. In order to explain the internal mechanism of the risk model in predicting the efficacy of immunotherapy, we analyzed the differentially expressed genes related to epithelial-mesenchymal transition (EMT) between two risk groups. We extracted RNA from normal bronchial epithelial cell and LUAD cells and verified the expression level of lncRNAs in the risk model by a quantitative real-time polymerase chain reaction (qRT-PCR) test. We compared our risk model with other published prognostic signatures with data from an independent cohort. We transfected LUAD cell with siRNA-LINC0253. Western blot analysis was performed to observed change of EMT-related marker in protein level.
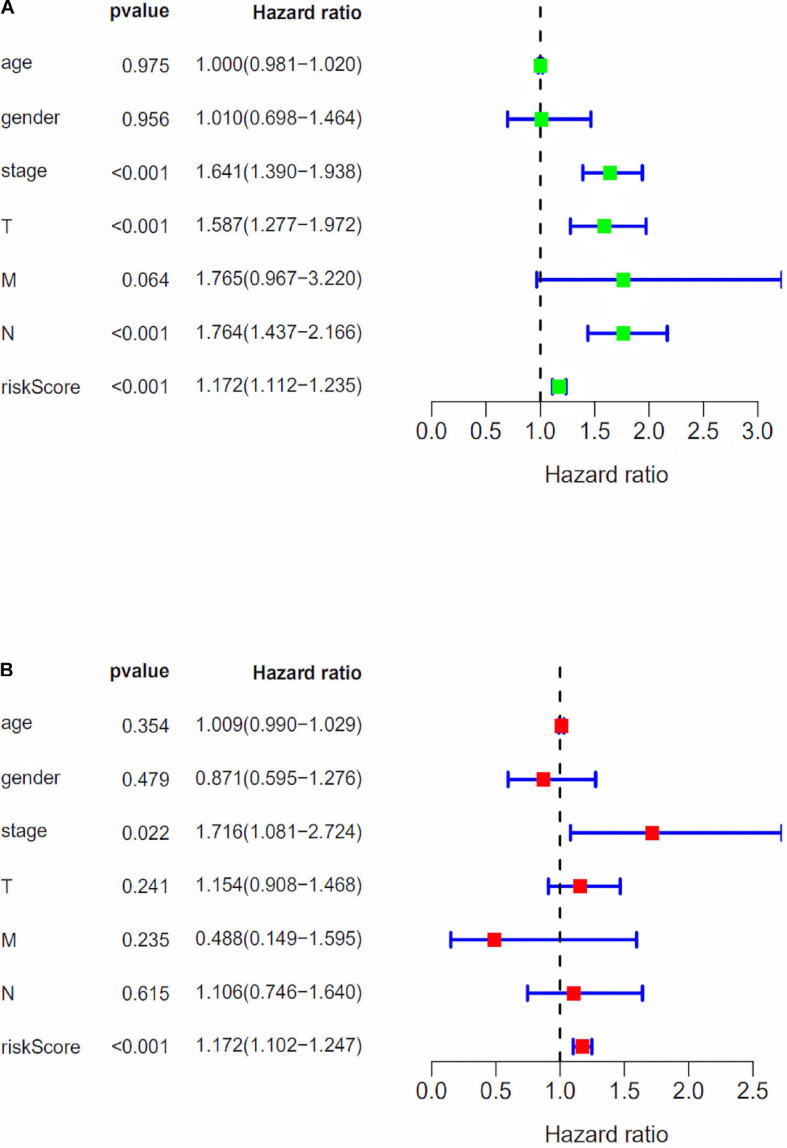
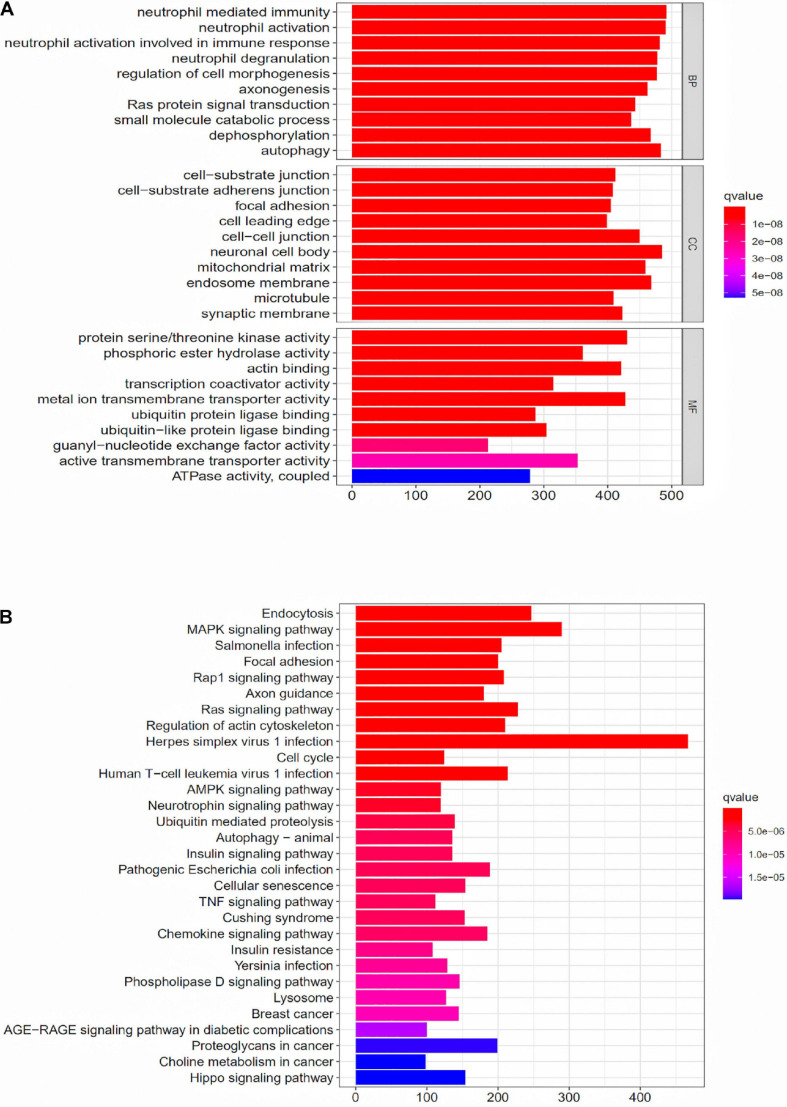
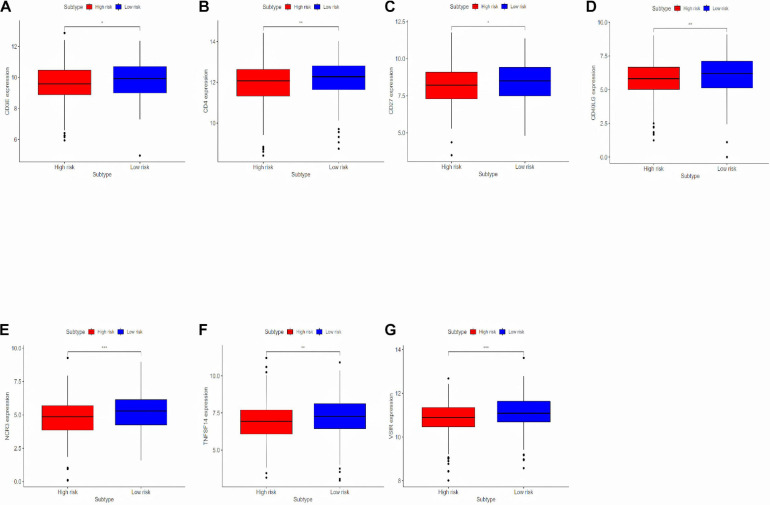
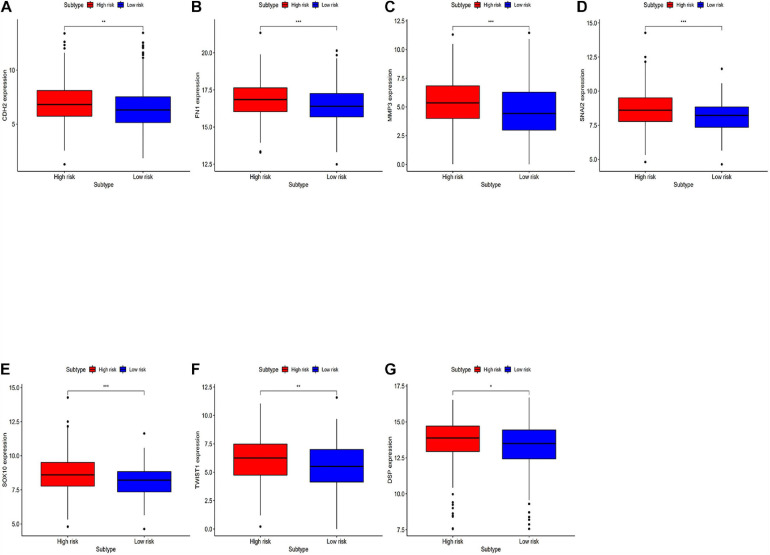
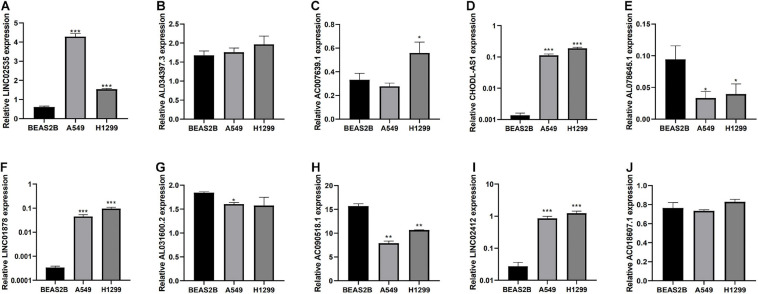
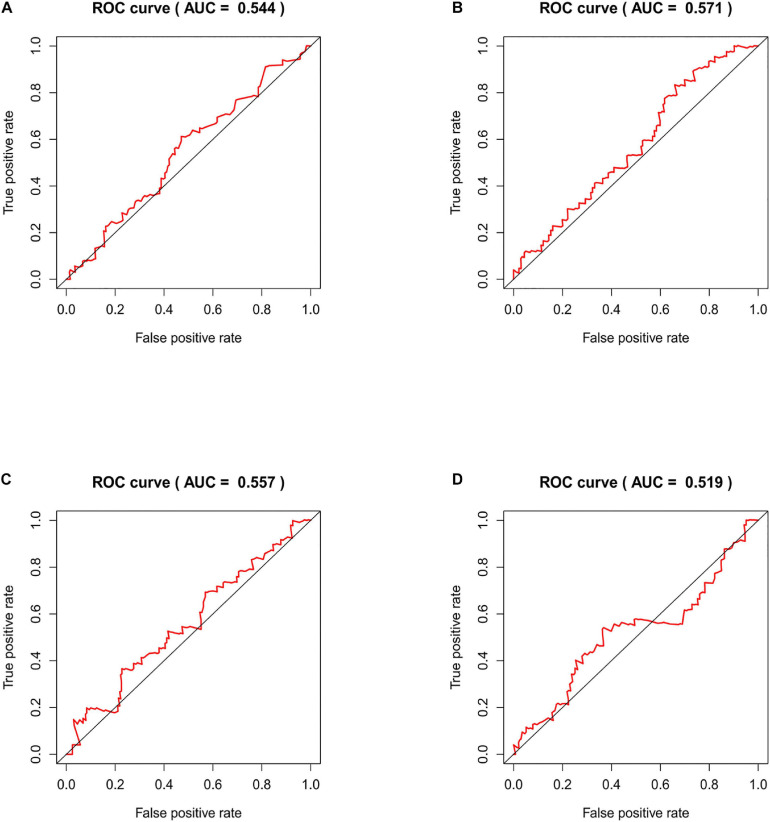
Through univariate Cox regression analysis, 24 immune-related lncRNAs were found to be strongly associated with the survival of the TCGA-LUAD dataset. Utilizing multivariate Cox regression analysis, 10 lncRNAs were selected to establish the risk model. The K-M survival curves and the ROC (AUC) curves proved that the risk model has a fine predictive effect. The GO enrichment analysis indicated that the effect of the differentially expressed genes between high-risk and low-risk groups is mainly involved in immune response and intercellular interaction. The KEGG enrichment analysis indicated that the differentially expressed genes between high-risk and low-risk groups are mainly involved in endocytosis and the MAPK signaling pathway. The expression of genes related to the efficacy of immunotherapy was significantly different between the two groups. A qRT-PCR test verified the expression level of lncRNAs in LUAD cells in the risk model. The AUC of ROC of 5 years in the independent validation dataset showed that this model had superior accuracy. Western blot analysis verified the change of EMT-related marker in protein level.
The immune lncRNA risk model established by us could better predict the prognosis of patients with LUAD.
肺腺癌(LUAD)主要起源于支气管的黏液上皮和腺上皮。它是非小细胞肺癌(NSCLC)最常见的病理亚型。目前,仍缺乏明确的标准来预测免疫治疗的疗效。LUAD患者的5年生存率仍然较低。
所有数据均从癌症基因组图谱(TCGA)数据库下载。我们使用基因集富集分析(GSEA)数据库获取免疫相关的mRNA。通过使用R 3.6.3版本(Pearson相关系数cor = 0.5,P < 0.05)对免疫相关基因进行相关性检验来获取免疫相关的lncRNA。将TCGA-LUAD数据集随机分为测试集和训练集。基于训练集进行单变量和多变量Cox回归分析,我们筛选出预后免疫相关lncRNA并为每个样本给出一个风险评分。根据中位风险评分将样本分为高风险组和低风险组。通过结合Kaplan-Meier(KM)生存曲线、受试者工作特征(ROC)(AUC)曲线、独立危险因素分析以及样本的临床数据,我们评估了风险模型的准确性。对高风险组和低风险组之间差异表达的mRNA进行基因本体(GO)富集分析和京都基因与基因组百科全书(KEGG)富集分析。分析两个风险组之间与免疫反应相关的差异表达基因,以评估该模型在预测免疫治疗疗效和效果方面的作用。为了解释风险模型在预测免疫治疗疗效中的内在机制,我们分析了两个风险组之间与上皮-间质转化(EMT)相关的差异表达基因。我们从正常支气管上皮细胞和LUAD细胞中提取RNA,并通过定量实时聚合酶链反应(qRT-PCR)试验验证风险模型中lncRNA的表达水平。我们将我们的风险模型与其他已发表的预后特征与来自独立队列的数据进行比较。我们用siRNA-LINC0253转染LUAD细胞。进行蛋白质印迹分析以观察EMT相关标志物在蛋白质水平的变化。
通过单变量Cox回归分析,发现24个免疫相关lncRNA与TCGA-LUAD数据集的生存密切相关。利用多变量Cox回归分析,选择10个lncRNA建立风险模型。K-M生存曲线和ROC(AUC)曲线证明该风险模型具有良好的预测效果。GO富集分析表明,高风险组和低风险组之间差异表达基因的作用主要涉及免疫反应和细胞间相互作用。KEGG富集分析表明,高风险组和低风险组之间差异表达基因主要涉及内吞作用和MAPK信号通路。两组之间与免疫治疗疗效相关基因的表达存在显著差异。qRT-PCR试验验证了风险模型中LUAD细胞中lncRNA的表达水平。独立验证数据集中5年ROC的AUC表明该模型具有卓越的准确性。蛋白质印迹分析验证了EMT相关标志物在蛋白质水平的变化。
我们建立的免疫lncRNA风险模型可以更好地预测LUAD患者的预后。