Govindammal M, Prasath M, Kamaraj S, Muthu S, Selvapandiyan M
Department of Physics, Periyar University PG Extension Centre, Dharmapuri, 636701, India.
Department of Biotechnology, Periyar University PG Extension Centre, Dharmapuri, India.
Heliyon. 2021 Apr 7;7(4):e06646. doi: 10.1016/j.heliyon.2021.e06646. eCollection 2021 Apr.
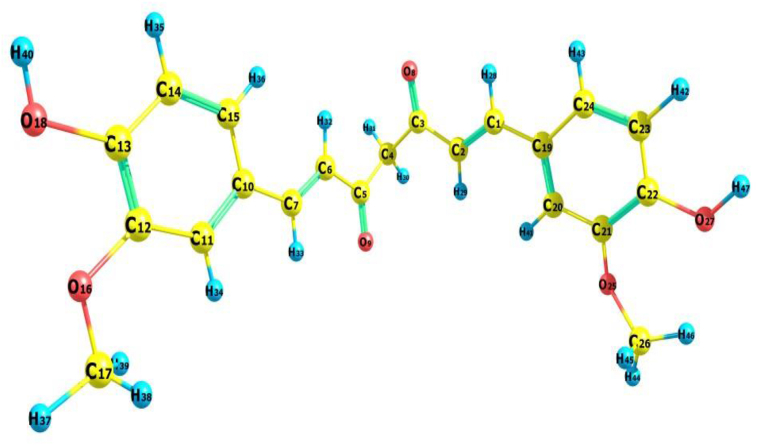
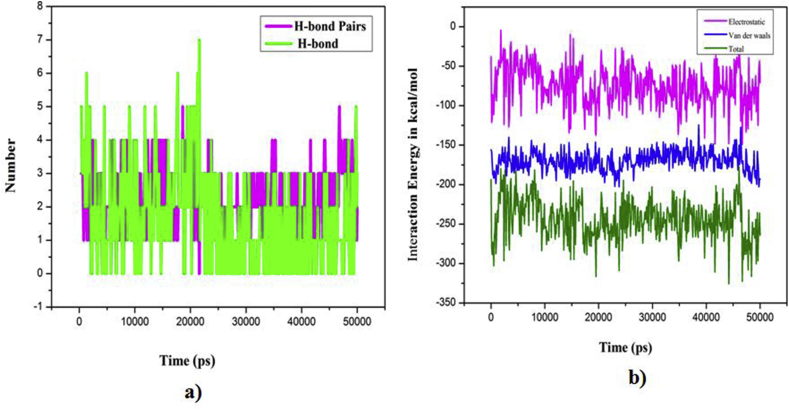
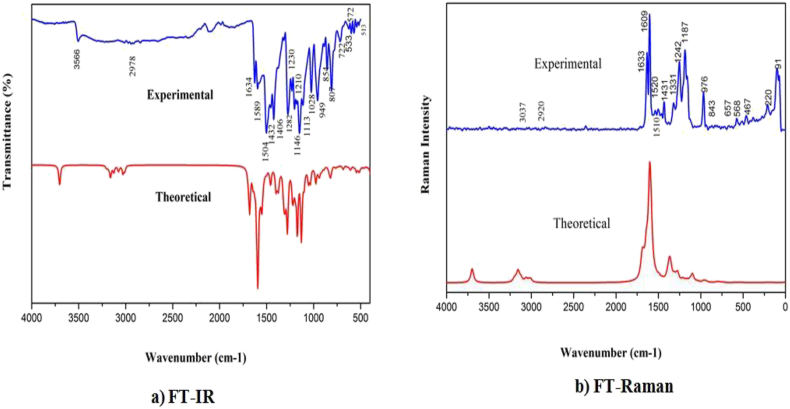
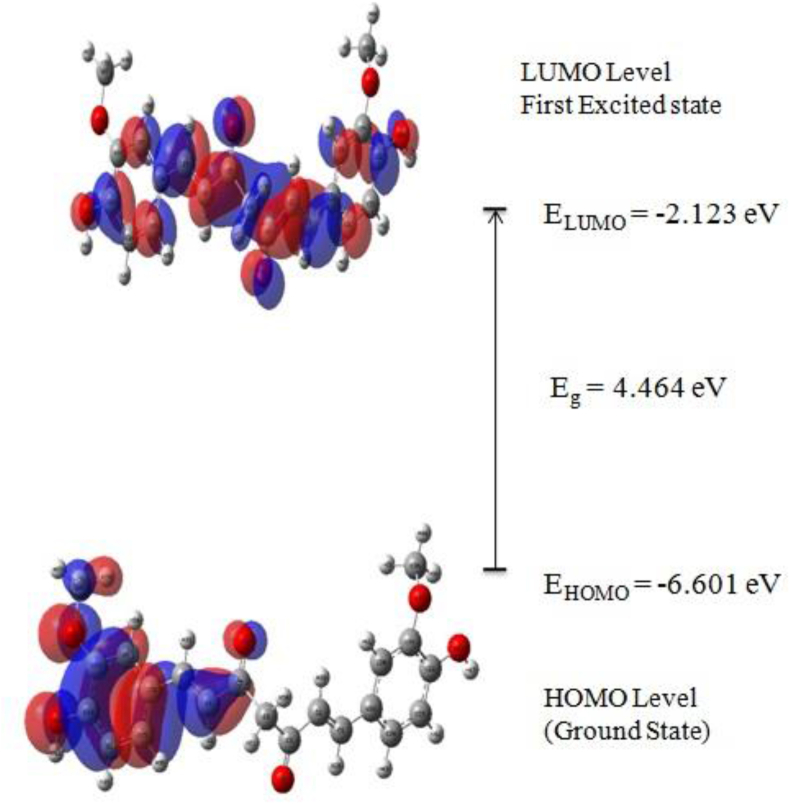
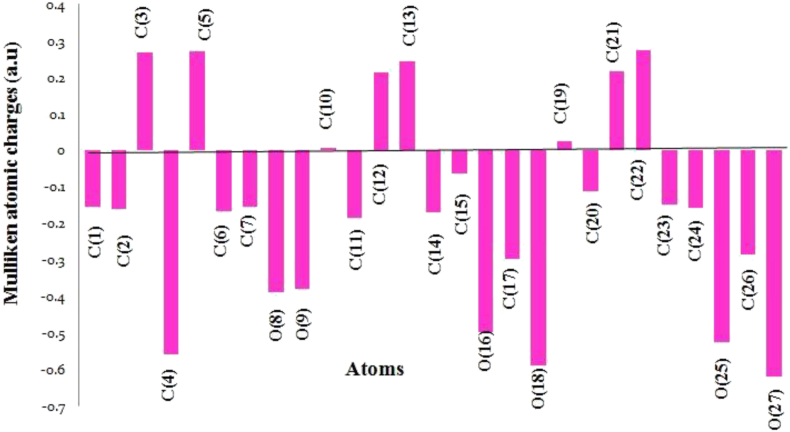
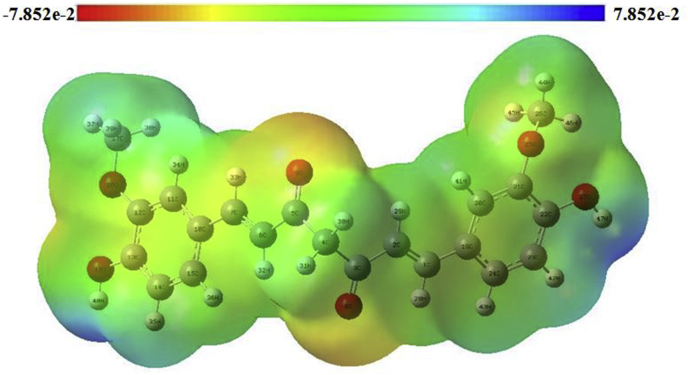
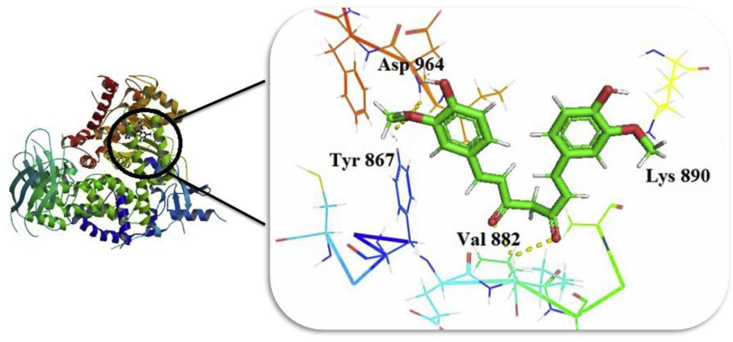
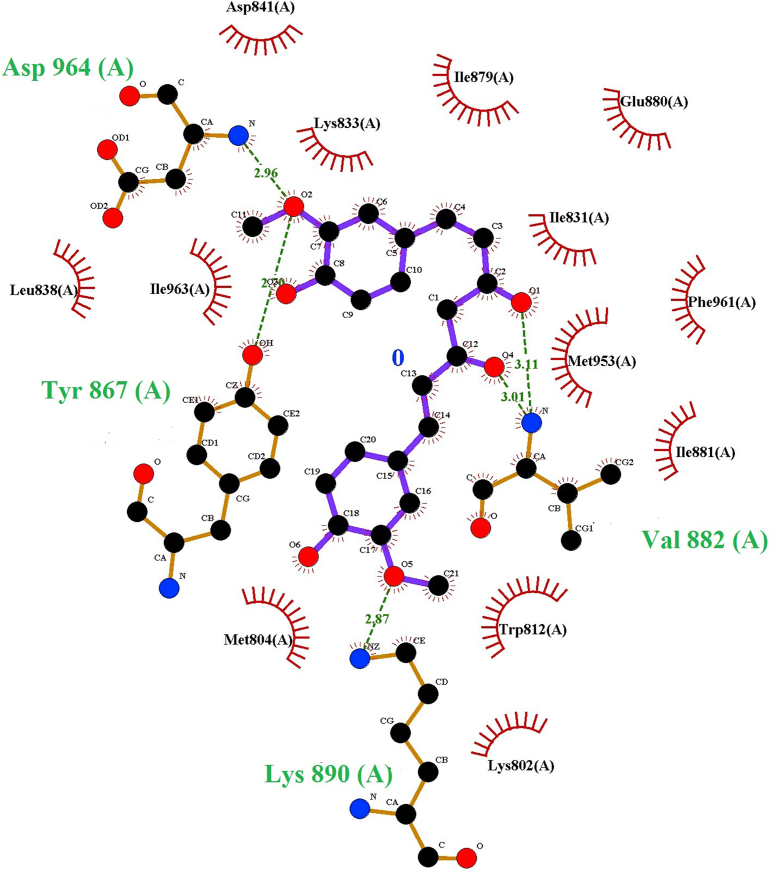
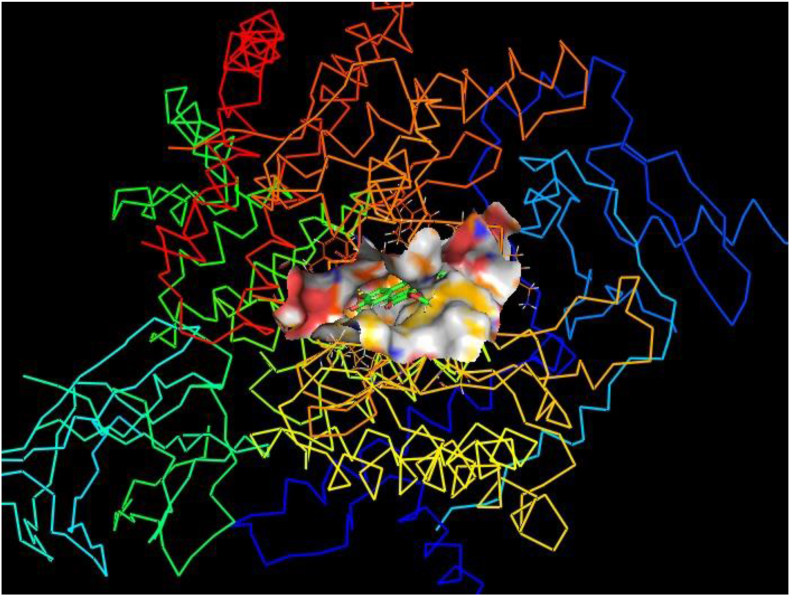
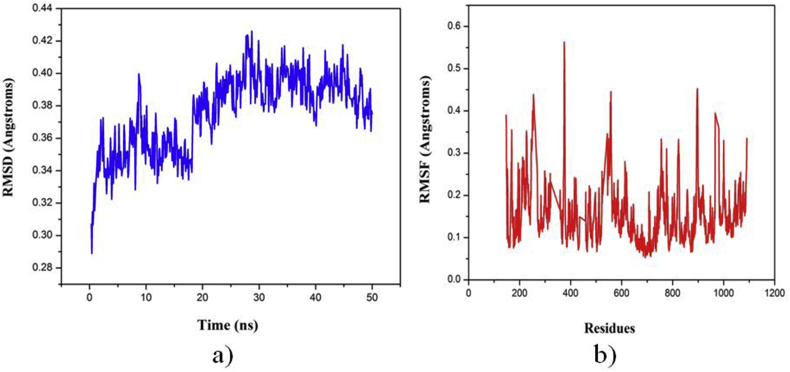
The IUPAC name of curcumin is (1E, 6E)-1,7-Bis(4-hydroxy-3methoxyphenyl) hepta-1,6-e-3,5-dione (7B3M5D) and is characterized by spectroscopic profiling with FT-IR and FT-Raman spectra obtained both experimentally and theoretically. PED analysis was done for the confirmation of minimum energy obtained in the title compound. Optimized geometrical parameters are compared with experimental values obtained for 7B3M5D by utilizing B3LYP functional employing 6-311++G (d,p) level of theory. The HOMO-LUMO, MEP, and Fukui function analysis has been used to elucidate the information regarding charge transfer within the molecule. The stabilization energy and charge delocalization of the 7B3M5D were performed by NBO analysis. This article assesses that the title compound act as a potential inhibitor of the PI3K/AKT inhibitor through in silico studies, like molecular docking, molecular dynamics (MD), ADMET prediction and also this molecule obeys Lipinski's rule of five. 7B3M5D was docked effectively in the active site of PI3K/AKT inhibitor.
姜黄素的国际纯粹与应用化学联合会(IUPAC)名称为(1E, 6E)-1,7-双(4-羟基-3-甲氧基苯基)庚-1,6-二烯-3,5-二酮(7B3M5D),其通过实验和理论获得的傅里叶变换红外光谱(FT-IR)和傅里叶变换拉曼光谱进行光谱分析表征。进行势能分布(PED)分析以确认标题化合物中获得的最低能量。利用采用6-311++G(d,p)理论水平的B3LYP泛函,将优化的几何参数与7B3M5D的实验值进行比较。已使用最高已占分子轨道-最低未占分子轨道(HOMO-LUMO)、分子静电势(MEP)和福井函数分析来阐明有关分子内电荷转移的信息。通过自然键轨道(NBO)分析对7B3M5D的稳定能和电荷离域进行了研究。本文通过计算机模拟研究评估标题化合物作为PI3K/AKT抑制剂的潜在抑制剂,如分子对接、分子动力学(MD)、药物代谢及毒性预测,并且该分子符合Lipinski的五规则。7B3M5D有效地对接在PI3K/AKT抑制剂的活性位点。