CNRS, LS2N, Université de Nantes, 44000, Nantes, France.
UR BIA, INRAE, 44316, Nantes, France.
BMC Bioinformatics. 2021 Apr 26;22(Suppl 2):65. doi: 10.1186/s12859-021-03963-6.
Mass spectrometry remains the privileged method to characterize proteins. Nevertheless, most of the spectra generated by an experiment remain unidentified after their analysis, mostly because of the modifications they carry. Open Modification Search (OMS) methods offer a promising answer to this problem. However, assessing the quality of OMS identifications remains a difficult task.
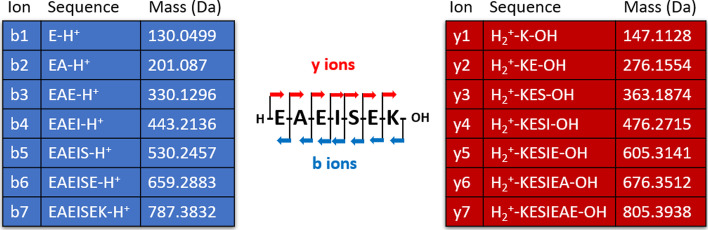
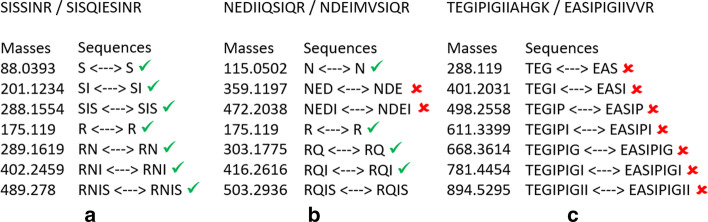
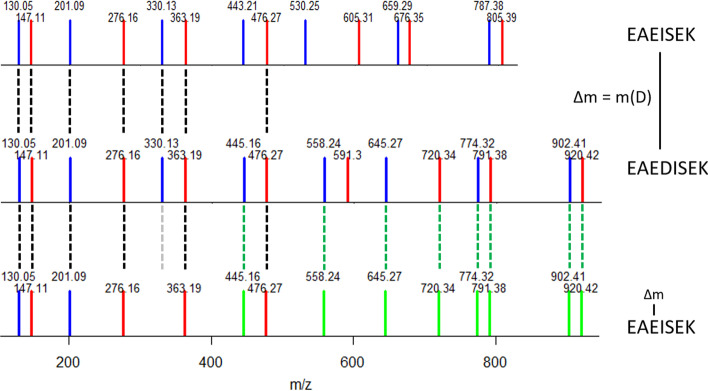
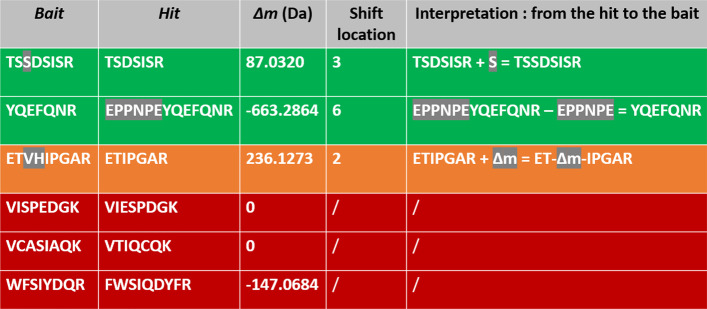
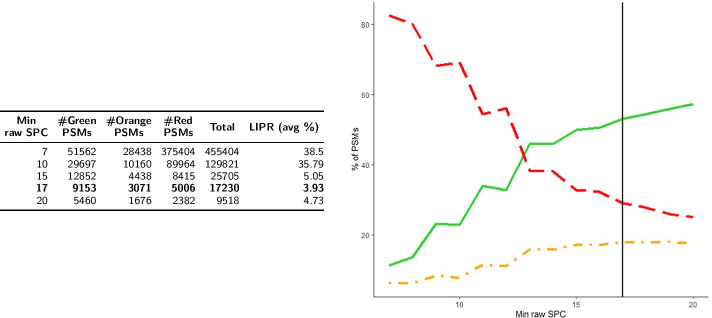
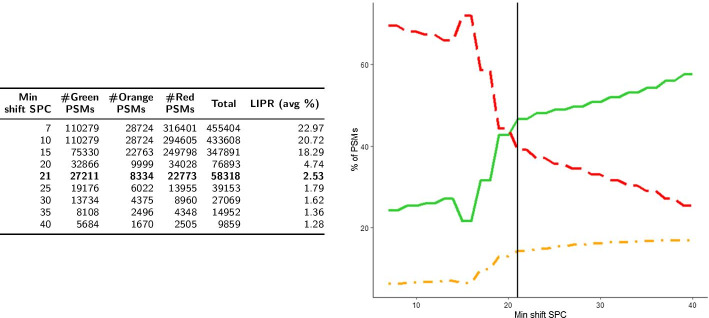
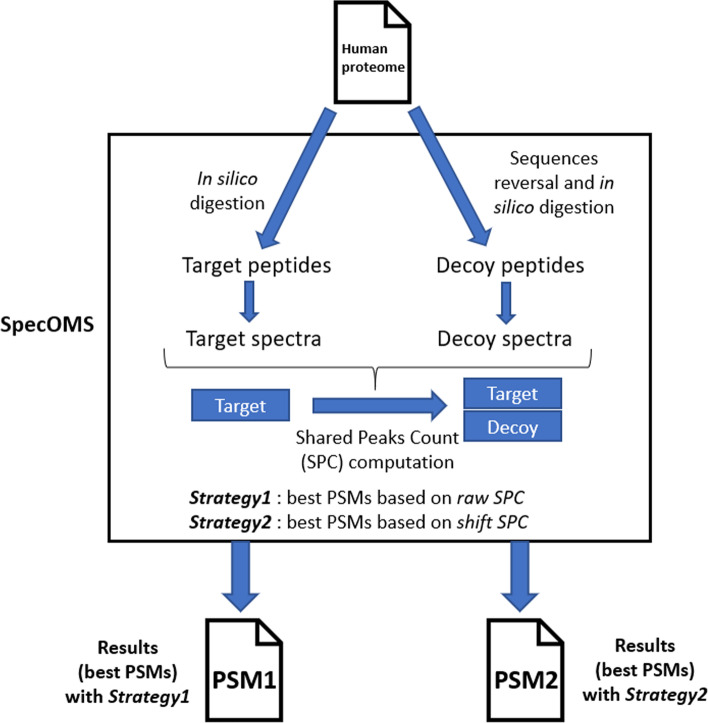
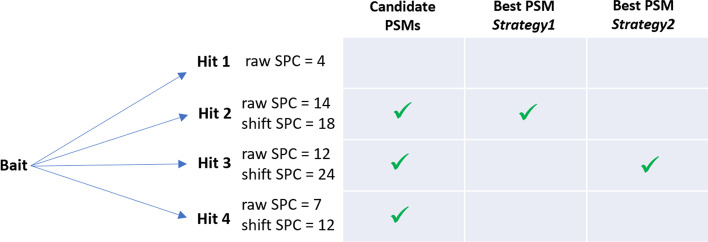
Aiming at better understanding the relationship between (1) similarity of pairs of spectra provided by OMS methods and (2) relevance of their corresponding peptide sequences, we used a dataset composed of theoretical spectra only, on which we applied two OMS strategies. We also introduced two appropriately defined measures for evaluating the above mentioned spectra/sequence relevance in this context: one is a color classification representing the level of difficulty to retrieve the proper sequence of the peptide that generated the identified spectrum ; the other, called LIPR, is the proportion of common masses, in a given Peptide Spectrum Match (PSM), that represent dissimilar sequences. These two measures were also considered in conjunction with the False Discovery Rate (FDR).
According to our measures, the strategy that selects the best candidate by taking the mass difference between two spectra into account yields better quality results. Besides, although the FDR remains an interesting indicator in OMS methods (as shown by LIPR), it is questionable: indeed, our color classification shows that a non negligible proportion of relevant spectra/sequence interpretations corresponds to PSMs coming from the decoy database.
The three above mentioned measures allowed us to clearly determine which of the two studied OMS strategies outperformed the other, both in terms of number of identifications and of accuracy of these identifications. Even though quality evaluation of PSMs in OMS methods remains challenging, the study of theoretical spectra is a favorable framework for going further in this direction.
质谱仍然是鉴定蛋白质的首选方法。然而,大多数实验生成的光谱在分析后仍然无法识别,主要是因为它们携带的修饰。开放修饰搜索 (OMS) 方法为此问题提供了一个有前途的答案。然而,评估 OMS 鉴定的质量仍然是一项艰巨的任务。
为了更好地理解 (1) OMS 方法提供的一对光谱之间的相似性和 (2) 它们对应的肽序列的相关性,我们仅使用理论光谱数据集,在该数据集中应用了两种 OMS 策略。我们还引入了两个适当定义的度量标准来评估上述光谱/序列相关性:一个是颜色分类,代表检索生成鉴定光谱的肽的正确序列的难度级别;另一个称为 LIPR,是给定肽谱匹配 (PSM) 中代表不同序列的共同质量的比例。这两个度量标准也与错误发现率 (FDR) 一起考虑。
根据我们的度量标准,考虑两个光谱之间质量差异来选择最佳候选的策略产生了更好质量的结果。此外,尽管 FDR 仍然是 OMS 方法中的一个有趣指标(如 LIPR 所示),但它值得怀疑:实际上,我们的颜色分类表明,大量相关的光谱/序列解释对应于来自诱饵数据库的 PSM。
上述三个度量标准使我们能够清楚地确定两种研究 OMS 策略中的哪一种优于另一种,无论是在鉴定数量还是鉴定准确性方面。尽管 OMS 方法中 PSM 的质量评估仍然具有挑战性,但对理论光谱的研究是朝着这个方向进一步发展的有利框架。