Department of Mathematics and Computer Science , University of Antwerp , 2020 Antwerp , Belgium.
Biomedical Informatics Network Antwerpen (biomina) , 2020 Antwerp , Belgium.
J Proteome Res. 2019 Oct 4;18(10):3792-3799. doi: 10.1021/acs.jproteome.9b00291. Epub 2019 Aug 30.
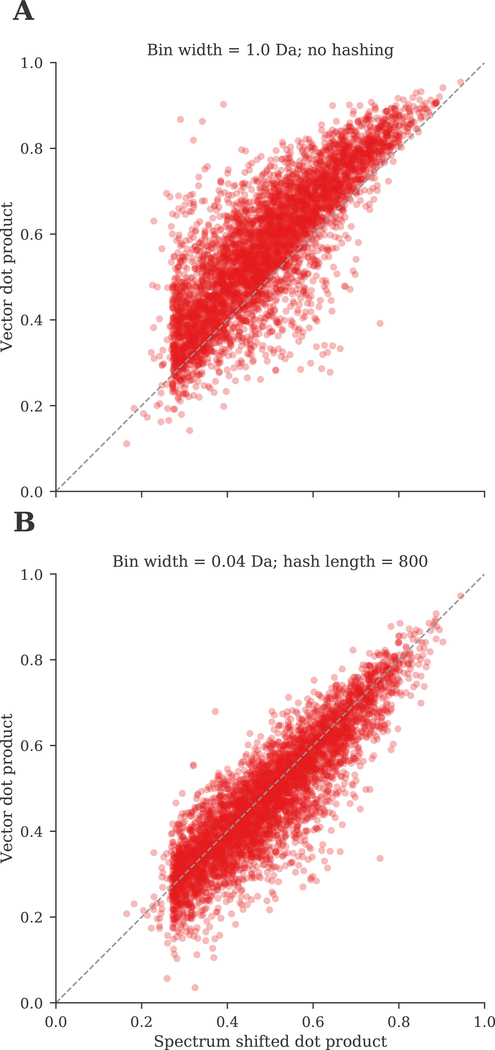
Open modification searching (OMS) is a powerful search strategy to identify peptides with any type of modification. OMS works by using a very wide precursor mass window to allow modified spectra to match against their unmodified variants, after which the modification types can be inferred from the corresponding precursor mass differences. A disadvantage of this strategy, however, is the large computational cost, because each query spectrum has to be compared against a multitude of candidate peptides. We have previously introduced the ANN-SoLo tool for fast and accurate open spectral library searching. ANN-SoLo uses approximate nearest neighbor indexing to speed up OMS by selecting only a limited number of the most relevant library spectra to compare to an unknown query spectrum. Here we demonstrate how this candidate selection procedure can be further optimized using graphics processing units. Additionally, we introduce a feature hashing scheme to convert high-resolution spectra to low-dimensional vectors. On the basis of these algorithmic advances, along with low-level code optimizations, the new version of ANN-SoLo is up to an order of magnitude faster than its initial version. This makes it possible to efficiently perform open searches on a large scale to gain a deeper understanding about the protein modification landscape. We demonstrate the computational efficiency and identification performance of ANN-SoLo based on a large data set of the draft human proteome. ANN-SoLo is implemented in Python and C++. It is freely available under the Apache 2.0 license at https://github.com/bittremieux/ANN-SoLo .
开放修饰搜索(Open modification searching,OMS)是一种强大的搜索策略,可用于识别具有任何类型修饰的肽。OMS 通过使用非常宽的前体质量窗口来实现,允许修饰后的谱与未修饰的变体匹配,然后可以从相应的前体质量差异推断出修饰类型。然而,这种策略的一个缺点是计算成本高,因为每个查询谱都必须与大量候选肽进行比较。我们之前介绍了用于快速准确的开放光谱库搜索的 ANN-SoLo 工具。ANN-SoLo 使用近似最近邻索引来通过仅选择数量有限的最相关的库谱与未知查询谱进行比较来加速 OMS。在这里,我们展示了如何使用图形处理单元进一步优化这种候选选择过程。此外,我们引入了一种特征哈希方案,将高分辨率谱转换为低维向量。基于这些算法上的改进以及低级代码优化,新版本的 ANN-SoLo 的速度比其初始版本快一个数量级。这使得可以有效地进行大规模的开放搜索,以更深入地了解蛋白质修饰景观。我们基于人类蛋白质组草案的大型数据集展示了 ANN-SoLo 的计算效率和识别性能。ANN-SoLo 是用 Python 和 C++实现的。它在 Apache 2.0 许可证下可在 https://github.com/bittremieux/ANN-SoLo 上免费获得。