Department of Chemistry, University of Kansas, Lawrence, Kansas 66045, United States.
J Proteome Res. 2021 May 7;20(5):2823-2829. doi: 10.1021/acs.jproteome.1c00066. Epub 2021 Apr 28.
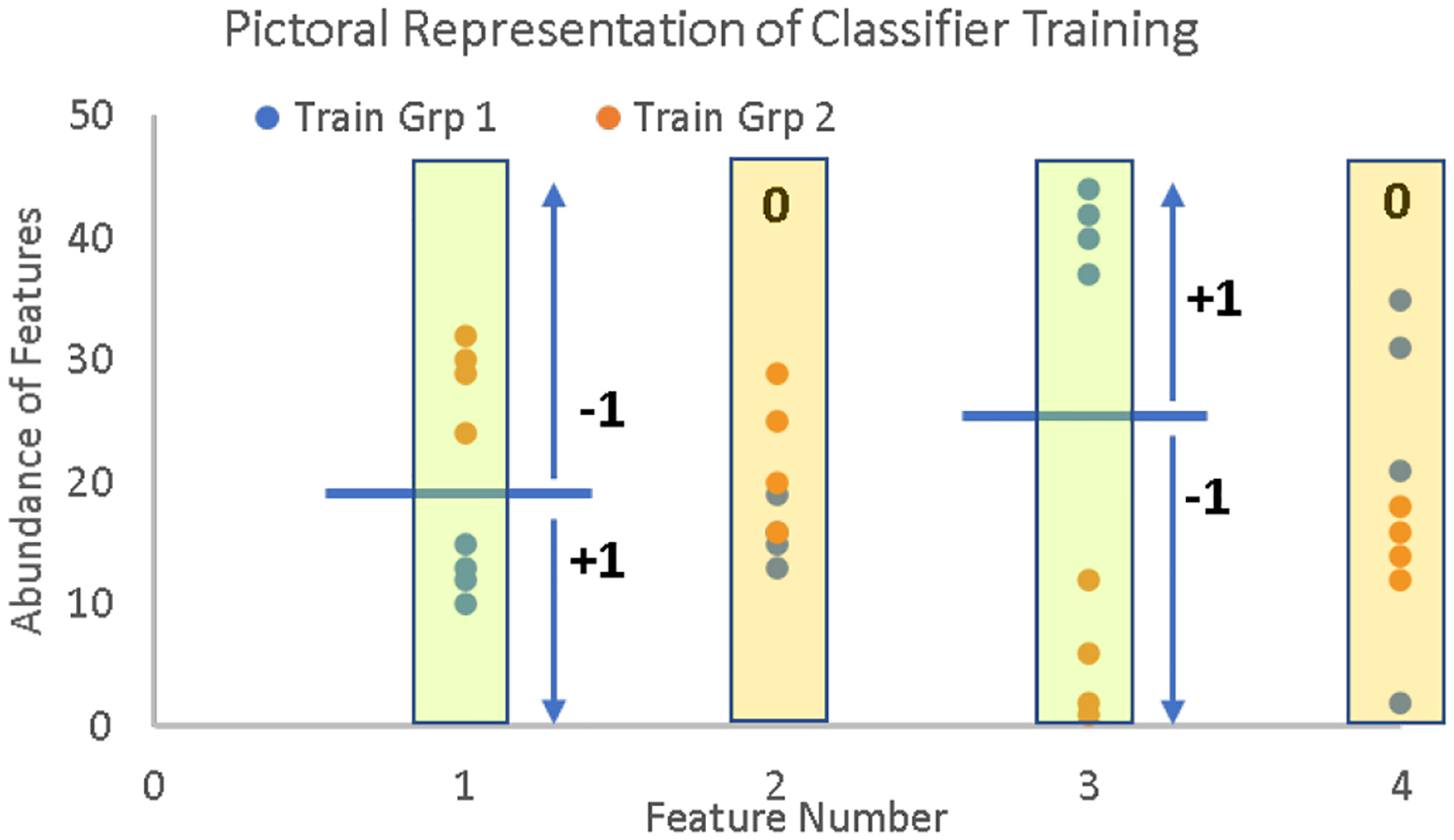
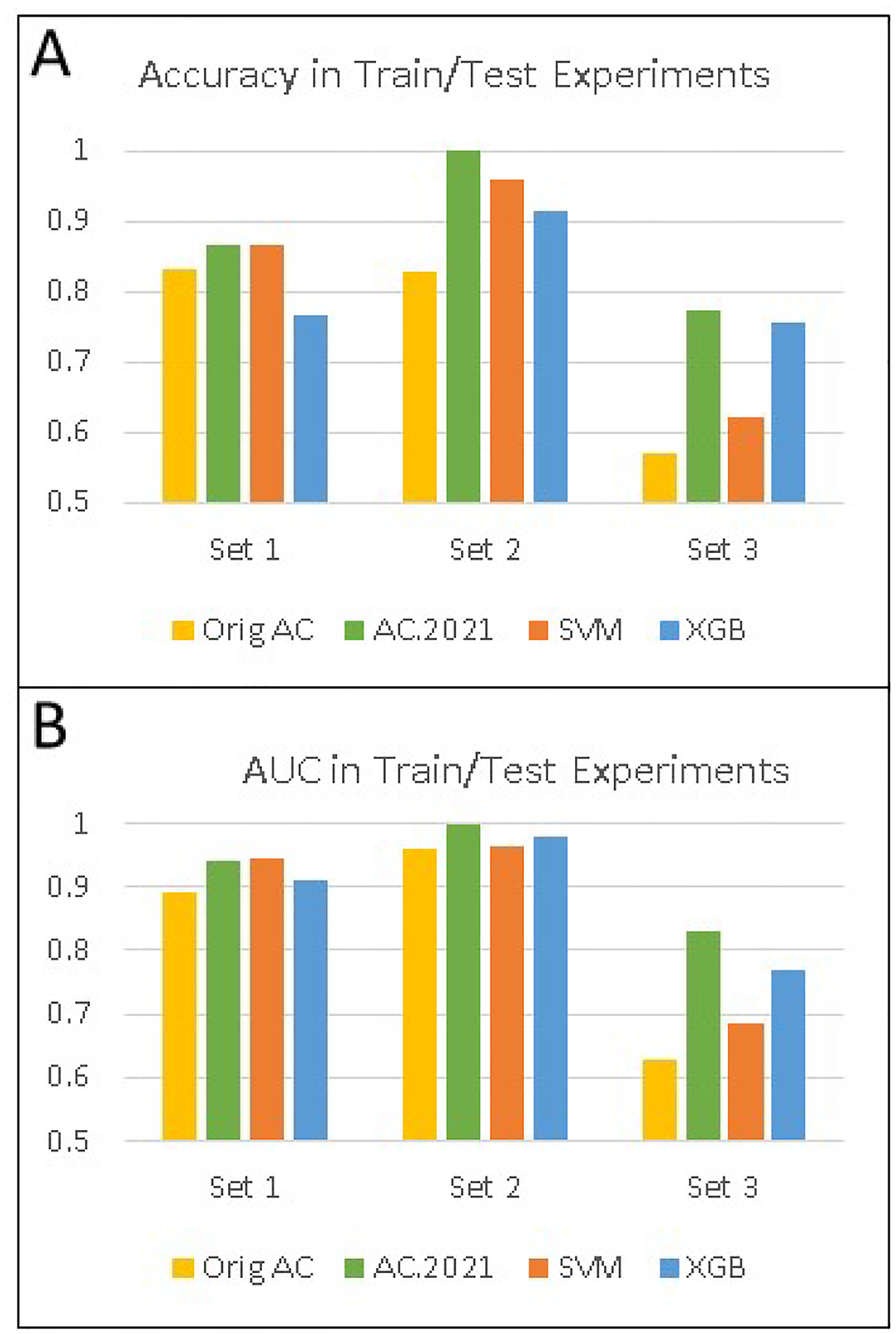
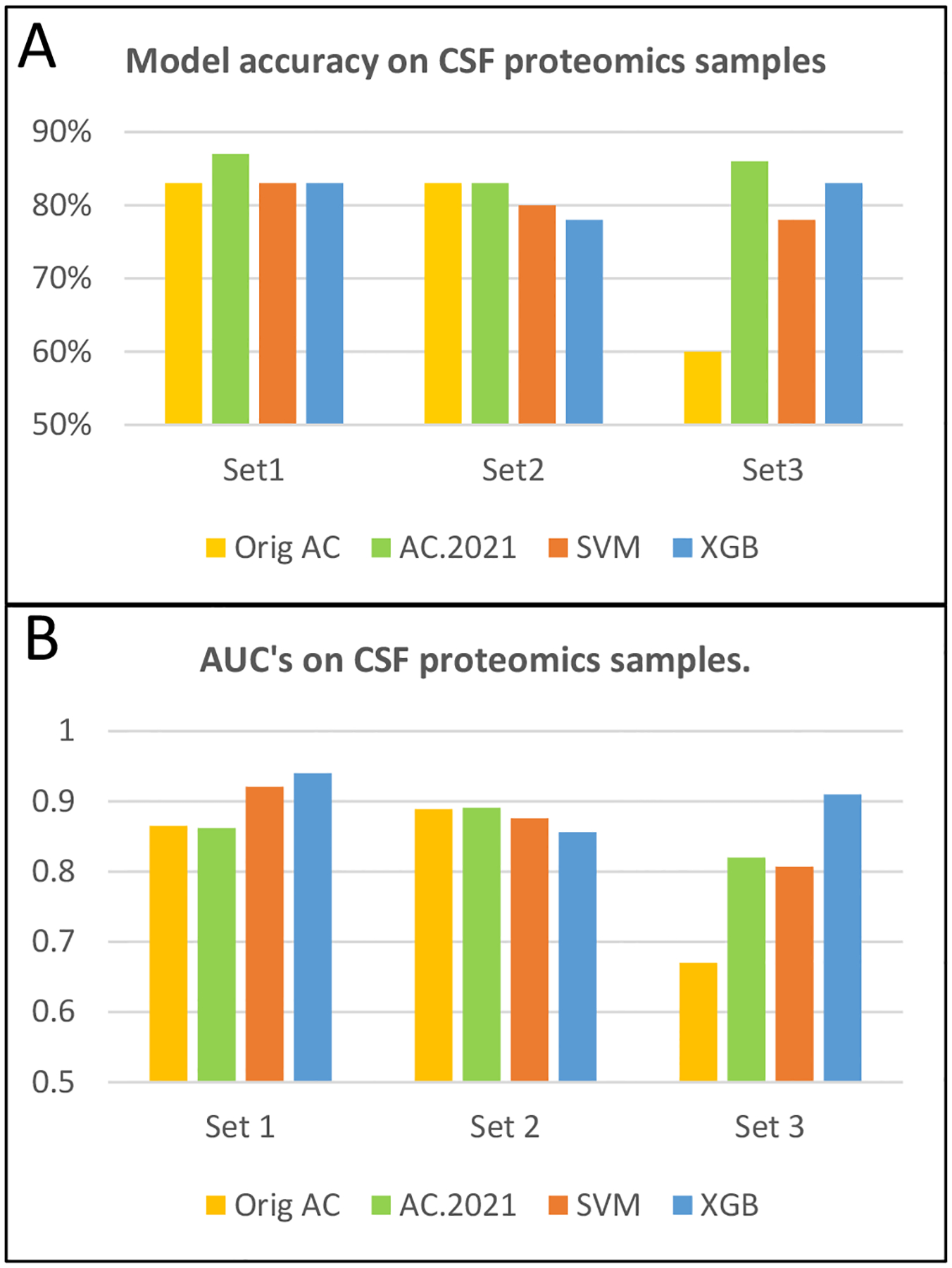
Mass spectrometry data sets from omics studies are an optimal information source for discriminating patients with disease and identifying biomarkers. Thousands of proteins or endogenous metabolites can be queried in each analysis, spanning several orders of magnitude in abundance. Machine learning tools that effectively leverage these data to accurately identify disease states are in high demand. While mass spectrometry data sets are rich with potentially useful information, using the data effectively can be challenging because of missing entries in the data sets and because the number of samples is typically much smaller than the number of features, two challenges that make machine learning difficult. To address this problem, we have modified a new supervised classification tool, the Aristotle Classifier, so that omics data sets can be better leveraged for identifying disease states. The optimized classifier, AC.2021, is benchmarked on multiple data sets against its predecessor and two leading supervised classification tools, Support Vector Machine (SVM) and XGBoost. The new classifier, AC.2021, outperformed existing tools on multiple tests using proteomics data. The underlying code for the classifier, provided herein, would be useful for researchers who desire improved classification accuracy when using their omics data sets to identify disease states.
组学研究的质谱数据集是区分疾病患者和识别生物标志物的最佳信息来源。在每次分析中可以查询数千种蛋白质或内源性代谢物,其丰度跨越几个数量级。能够有效利用这些数据准确识别疾病状态的机器学习工具需求量很大。虽然质谱数据集富含潜在有用的信息,但由于数据集存在缺失项,并且样本数量通常远小于特征数量,这两个挑战使得机器学习变得困难,因此有效地使用这些数据具有挑战性。为了解决这个问题,我们修改了一种新的有监督分类工具,即亚里士多德分类器,以便更好地利用组学数据集来识别疾病状态。优化后的分类器 AC.2021 在多个数据集上与前代产品以及两种领先的有监督分类工具支持向量机(SVM)和 XGBoost 进行了基准测试。新的分类器 AC.2021 在使用蛋白质组学数据进行的多项测试中均优于现有工具。此处提供的分类器的基础代码对于希望在使用组学数据集识别疾病状态时提高分类准确性的研究人员很有用。