Routh Shreya, Acharyya Anamika, Dhar Riddhiman
Department of Biotechnology, Indian Institute of Technology Kharagpur, Kharagpur 721302, West Bengal, India.
Biol Methods Protoc. 2021 Apr 2;6(1):bpab007. doi: 10.1093/biomethods/bpab007. eCollection 2021.
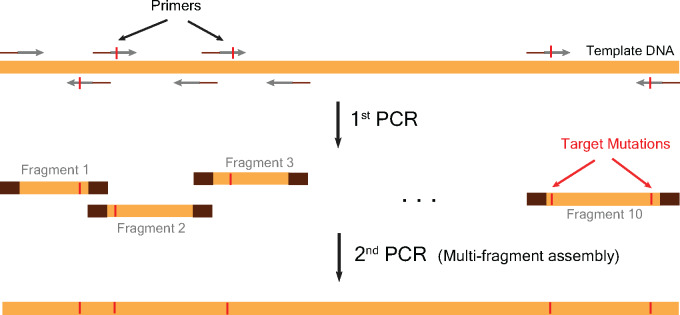
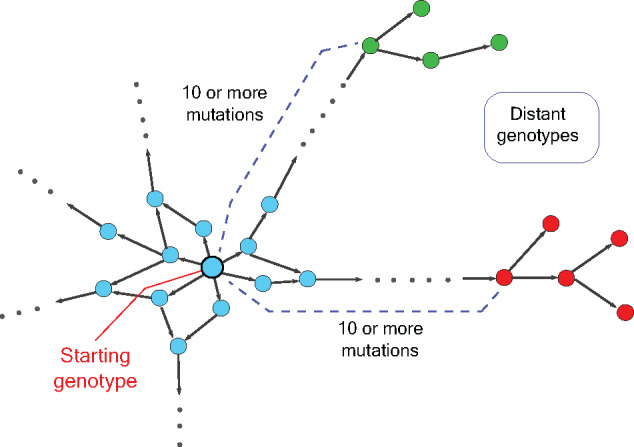
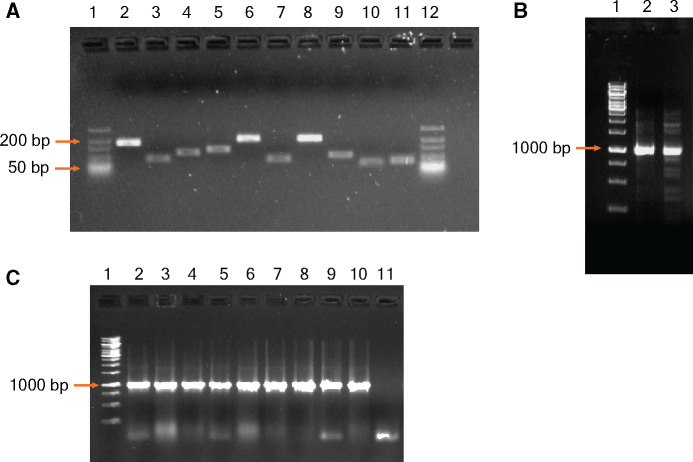
Construction of empirical fitness landscapes has transformed our understanding of genotype-phenotype relationships across genes. However, most empirical fitness landscapes have been constrained to the local genotype neighbourhood of a gene primarily due to our limited ability to systematically construct genotypes that differ by a large number of mutations. Although a few methods have been proposed in the literature, these techniques are complex owing to several steps of construction or contain a large number of amplification cycles that increase chances of non-specific mutations. A few other described methods require amplification of the whole vector, thereby increasing the chances of vector backbone mutations that can have unintended consequences for study of fitness landscapes. Thus, this has substantially constrained us from traversing large mutational distances in the genotype network, thereby limiting our understanding of the interactions between multiple mutations and the role these interactions play in evolution of novel phenotypes. In the current work, we present a simple but powerful approach that allows us to systematically and accurately construct gene variants at large mutational distances. Our approach relies on building-up small fragments containing targeted mutations in the first step followed by assembly of these fragments into the complete gene fragment by polymerase chain reaction (PCR). We demonstrate the utility of our approach by constructing variants that differ by up to 11 mutations in a model gene. Our work thus provides an accurate method for construction of multi-mutant variants of genes and therefore will transform the studies of empirical fitness landscapes by enabling exploration of genotypes that are far away from a starting genotype.
经验性适应度景观的构建改变了我们对跨基因的基因型-表型关系的理解。然而,大多数经验性适应度景观主要局限于一个基因的局部基因型邻域,这主要是因为我们系统构建大量突变差异基因型的能力有限。尽管文献中已经提出了一些方法,但这些技术由于构建步骤多而复杂,或者包含大量增加非特异性突变几率的扩增循环。其他一些描述的方法需要扩增整个载体,从而增加了载体骨架突变的几率,而这些突变可能会对适应度景观的研究产生意想不到的后果。因此,这极大地限制了我们在基因型网络中跨越较大突变距离,从而限制了我们对多个突变之间相互作用以及这些相互作用在新表型进化中所起作用的理解。在当前的工作中,我们提出了一种简单而强大的方法,使我们能够系统且准确地构建大突变距离的基因变体。我们的方法首先依赖于构建包含靶向突变的小片段,然后通过聚合酶链反应(PCR)将这些片段组装成完整的基因片段。我们通过构建在一个模型基因中相差多达11个突变的变体来证明我们方法的实用性。因此,我们的工作提供了一种构建基因多突变变体的准确方法,从而将通过探索远离起始基因型的基因型来改变经验性适应度景观的研究。