Departamento de Mejora Genética Animal, INIA, Ctra. de La Coruña, km 7.5, 28040, Madrid, Spain.
Centro de Investigación Mariña, Universidade de Vigo, Departamento de Bioquímica, Genética E Inmunología, Campus de Vigo, 36310, Vigo, Spain.
Genet Sel Evol. 2021 May 1;53(1):42. doi: 10.1186/s12711-021-00635-0.
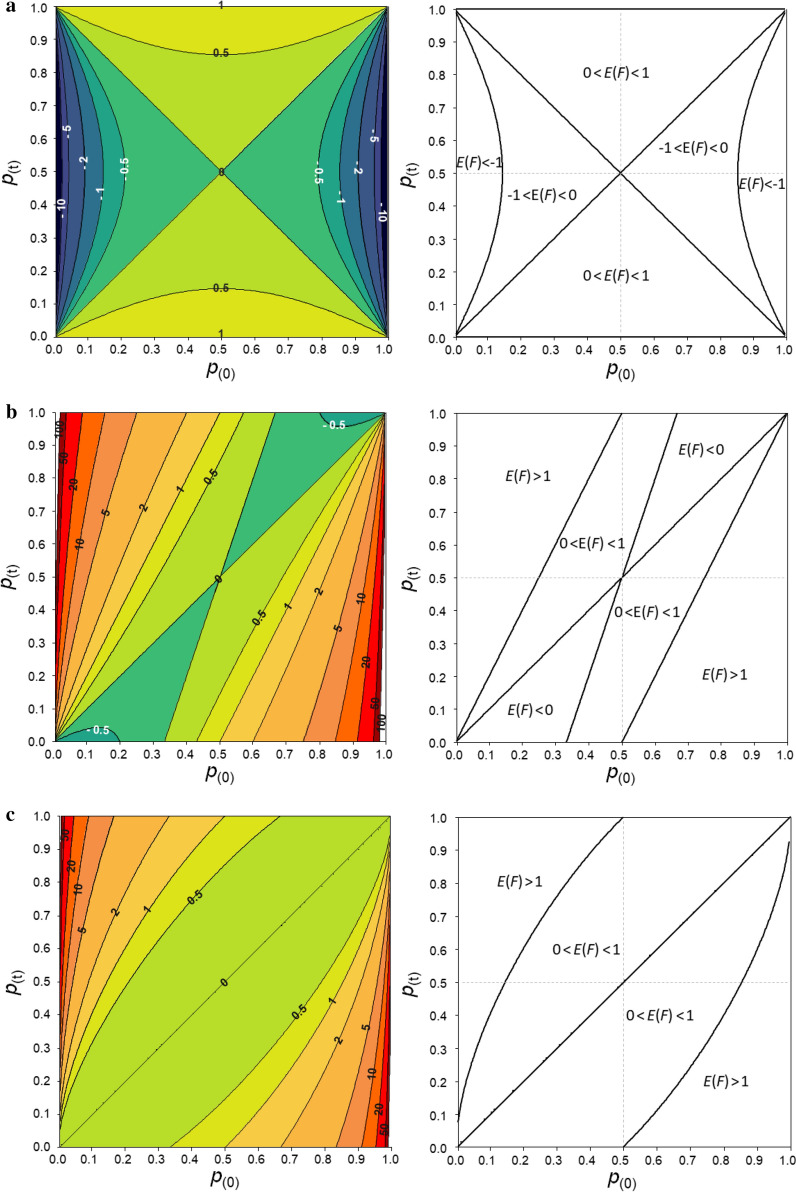
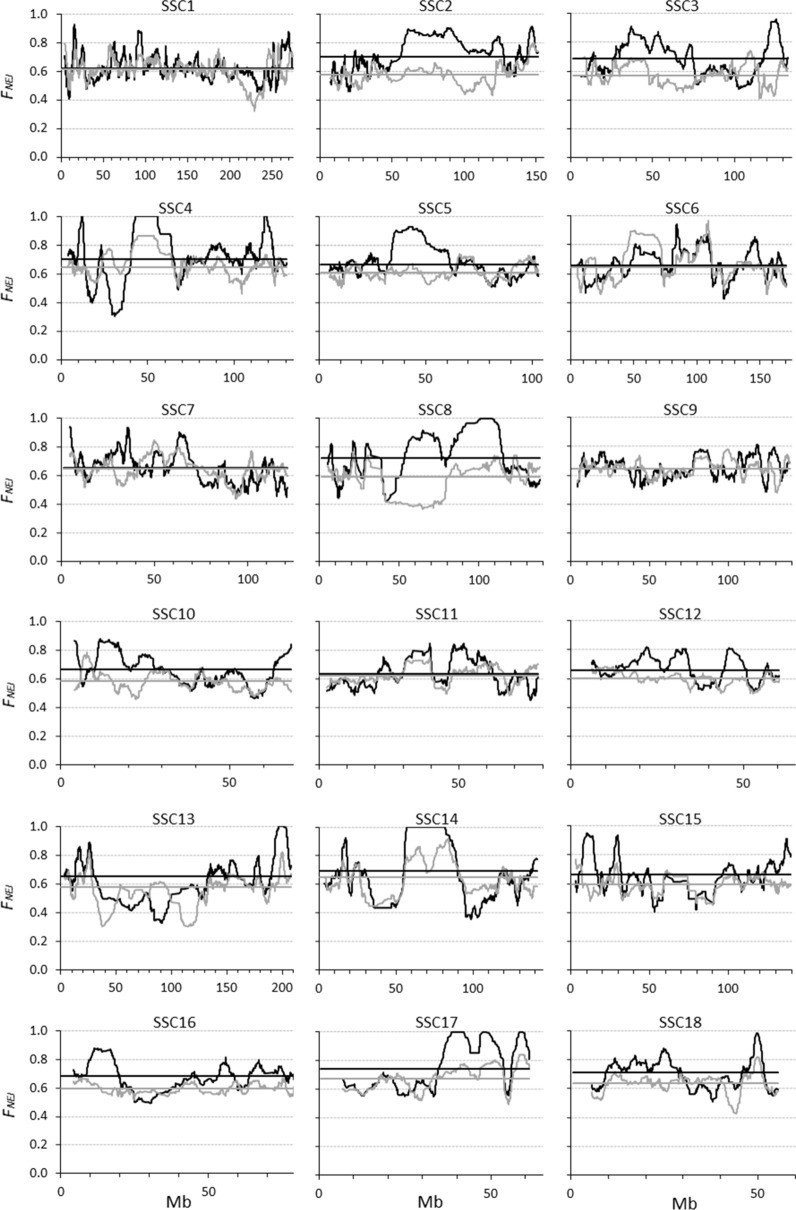
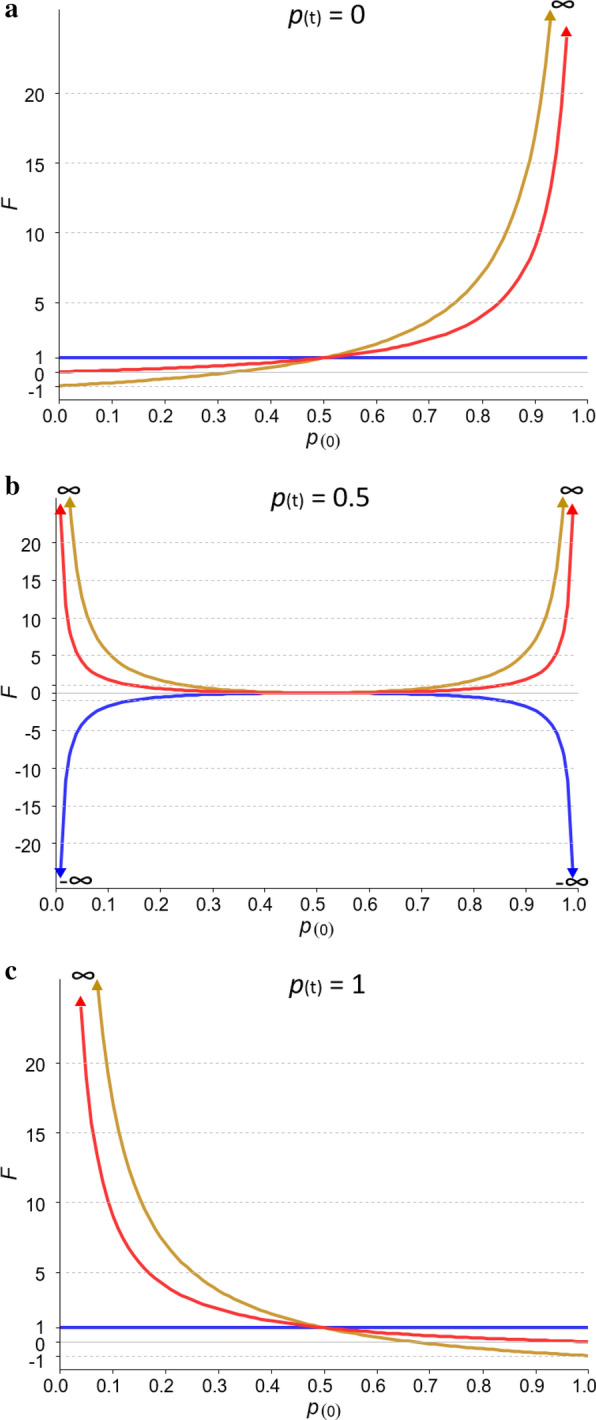
Genomic relationship matrices are used to obtain genomic inbreeding coefficients. However, there are several methodologies to compute these matrices and there is still an unresolved debate on which one provides the best estimate of inbreeding. In this study, we investigated measures of inbreeding obtained from five genomic matrices, including the Nejati-Javaremi allelic relationship matrix (F), the Li and Horvitz matrix based on excess of homozygosity (F), and the VanRaden (methods 1, F, and 2, F) and Yang (F) genomic relationship matrices. We derived expectations for each inbreeding coefficient, assuming a single locus model, and used these expectations to explain the patterns of the coefficients that were computed from thousands of single nucleotide polymorphism genotypes in a population of Iberian pigs.
Except for F, the evaluated measures of inbreeding do not match with the original definitions of inbreeding coefficient of Wright (correlation) or Malécot (probability). When inbreeding coefficients are interpreted as indicators of variability (heterozygosity) that was gained or lost relative to a base population, both F and F led to sensible results but this was not the case for F, F and F. When variability has increased relative to the base, F, F and F can indicate that it decreased. In fact, based on F, variability is not expected to increase. When variability has decreased, F and F can indicate that it has increased. Finally, these three coefficients can indicate that more variability than that present in the base population can be lost, which is also unreasonable. The patterns for these coefficients observed in the pig population were very different, following the derived expectations. As a consequence, the rate of inbreeding depression estimated based on these inbreeding coefficients differed not only in magnitude but also in sign.
Genomic inbreeding coefficients obtained from the diagonal elements of genomic matrices can lead to inconsistent results in terms of gain and loss of genetic variability and inbreeding depression estimates, and thus to misleading interpretations. Although these matrices have proven to be very efficient in increasing the accuracy of genomic predictions, they do not always provide a useful measure of inbreeding.
基因组关系矩阵用于获得基因组近交系数。然而,有几种方法可以计算这些矩阵,并且对于哪种方法提供最佳的近交估计值仍存在争议。在这项研究中,我们研究了从五个基因组矩阵中获得的近交系数度量,包括 Nejati-Javaremi 等位基因关系矩阵(F)、基于杂合过剩的 Li 和 Horvitz 矩阵(F)、VanRaden(方法 1、F 和 2、F)和 Yang(F)基因组关系矩阵。我们假设单一位点模型,推导出每个近交系数的期望,并使用这些期望来解释从伊比利亚猪群体中数千个单核苷酸多态性基因型计算得出的系数模式。
除了 F 之外,评估的近交系数与 Wright(相关)或 Malécot(概率)的原始近交系数定义不匹配。当近交系数被解释为相对于基础群体获得或失去的变异性(杂合性)的指标时,F 和 F 都导致了合理的结果,但 F、F 和 F 则不然。当变异性相对于基础群体增加时,F、F 和 F 可以表明它减少了。事实上,根据 F,变异性预计不会增加。当变异性减少时,F 和 F 可以表明它增加了。最后,这三个系数可以表明,比基础群体中存在的更多的变异性可能会丢失,这也是不合理的。在猪群体中观察到的这些系数的模式非常不同,与推导的期望一致。因此,基于这些近交系数估计的近交衰退率不仅在幅度上而且在符号上都不同。
从基因组矩阵的对角元素获得的基因组近交系数可能导致遗传变异性和近交衰退估计的增益和损失方面的不一致结果,并因此导致误导性解释。尽管这些矩阵已被证明在提高基因组预测的准确性方面非常有效,但它们并不总是提供有用的近交度量。