Jamal Qazi Mohammad Sajid, Ahmad Varish, Alharbi Ali H, Ansari Mohammad Azam, Alzohairy Mohammad A, Almatroudi Ahmad, Alghamdi Saad, Alomary Mohammad N, AlYahya Sami, Shesha Nashwa Talaat, Rehman Suriya
Department of Health Informatics, College of Public Health and Health Informatics, Qassim University, Al Bukayriyah, Saudi Arabia.
Health Information Technology Department, Faculty of Applied Studies, King Abdulaziz University, Jeddah, Saudi Arabia.
Saudi J Biol Sci. 2021 Aug;28(8):4560-4568. doi: 10.1016/j.sjbs.2021.04.057. Epub 2021 Apr 28.
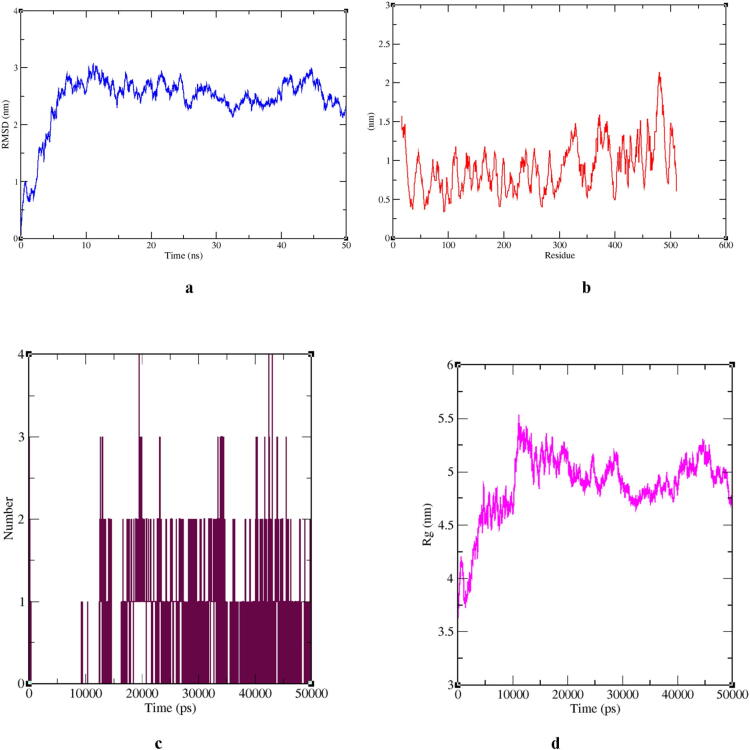
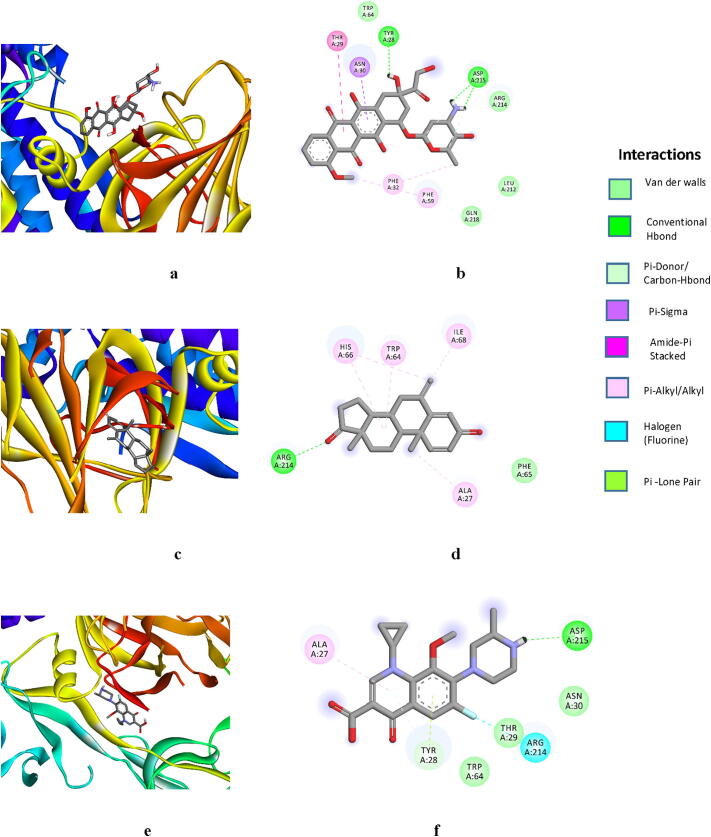
The human-to-human transmitted respiratory illness in COVID-19 affected by the pathogenic Severe Acute Respiratory Syndrome Corona Virus 2 (SARS-CoV-2), which appeared in the last of December 2019 in Wuhan, China, and rapidly spread in many countries. Thereon, based on the urgent need for therapeutic molecules, we conducted based docking and simulation molecular interaction studies on repurposing drugs, targeting SARS-CoV-2 spike protein. Further, the best binding energy of doxorubicin interacting with virus spike protein (PDB: 6VYB) was observed to be -6.38 and it was followed by exemestane and gatifloxacin. The molecular simulation dynamics analysis of doxorubicin, Reference Mean Square Deviation (RMSD), Root Mean Square fluctuation (RMSF), Radius of Gyration (Rg), and formation of hydrogen bonds plot interpretation suggested, a significant deviation and fluctuation of Doxorubicin-Spike RBD complex during the whole simulation period. The Rg analysis has stated that the Doxorubicin-Spike RBD complex was stable during 15,000-35,000 ps MDS. The results have suggested that doxorubicin could inhibit the virus spike protein and prevent the access of the SARS-CoV-2 to the host cell. Thus, research on these drugs could be advantageous to evaluate significant molecules that control the COVID-19 disease.
由致病性严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的2019冠状病毒病(COVID-19)是一种人传人的呼吸道疾病,于2019年12月底在中国武汉出现,并迅速在许多国家传播。因此,基于对治疗性分子的迫切需求,我们对用于治疗COVID-19的药物进行了分子对接和模拟分子相互作用研究,这些药物靶向SARS-CoV-2刺突蛋白。此外,观察到阿霉素与病毒刺突蛋白(PDB:6VYB)相互作用的最佳结合能为-6.38,其次是依西美坦和加替沙星。对阿霉素的分子模拟动力学分析、参考均方根偏差(RMSD)、均方根波动(RMSF)、回转半径(Rg)以及氢键形成图的解释表明,在整个模拟期间,阿霉素-刺突RBD复合物存在显著偏差和波动。Rg分析表明,在15,000-35,000皮秒的分子动力学模拟(MDS)过程中,阿霉素-刺突RBD复合物是稳定的。结果表明,阿霉素可以抑制病毒刺突蛋白,阻止SARS-CoV-2进入宿主细胞。因此,对这些药物的研究可能有利于评估控制COVID-19疾病的重要分子。