BCM Tailor Labs, Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, Texas, USA.
Infect Immun. 2021 Jul 15;89(8):e0011521. doi: 10.1128/IAI.00115-21.
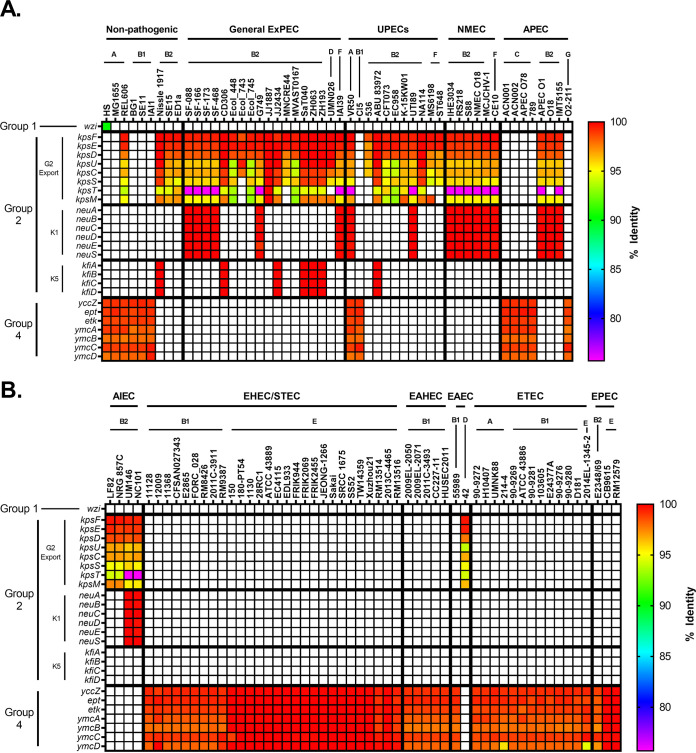
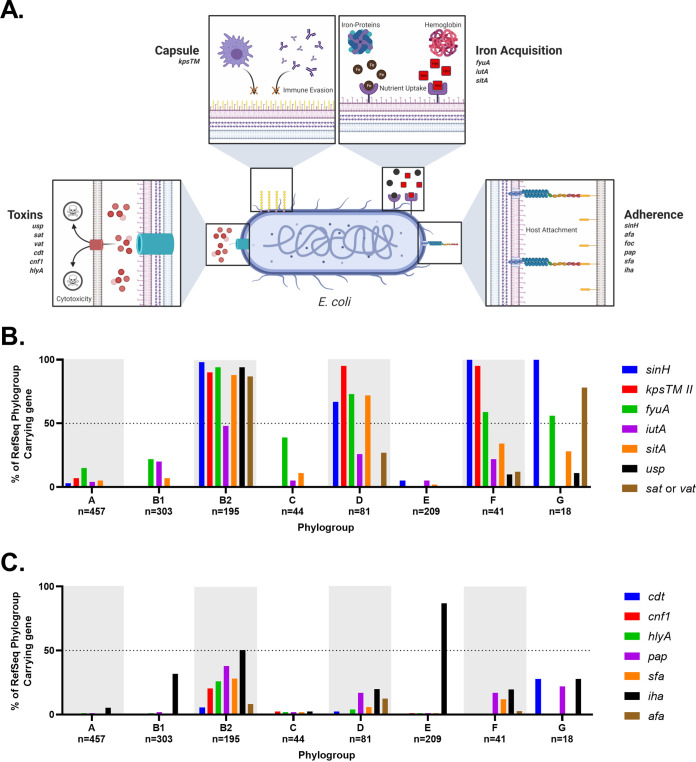
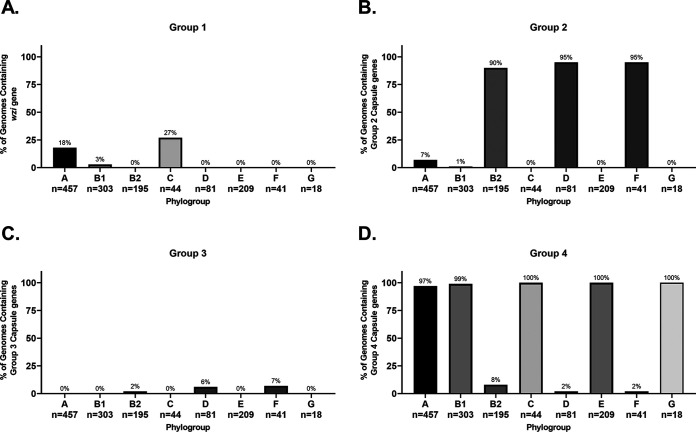
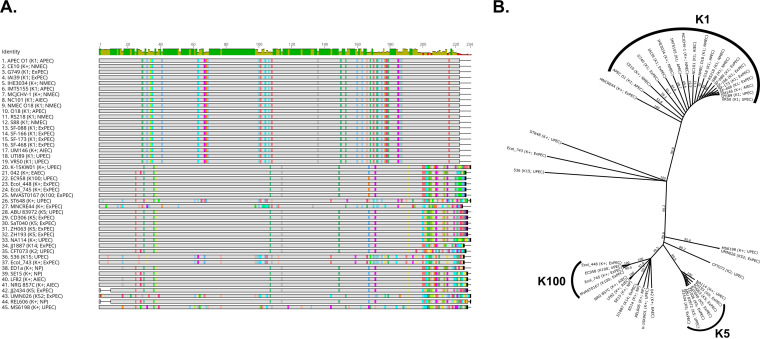
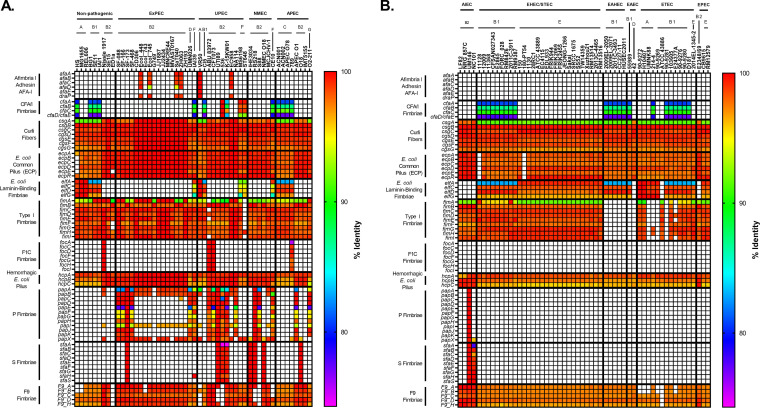
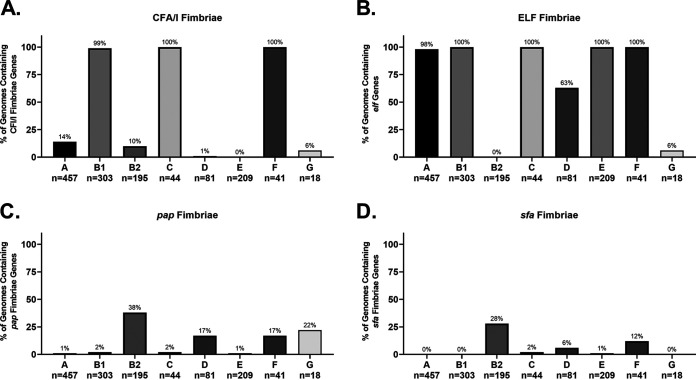
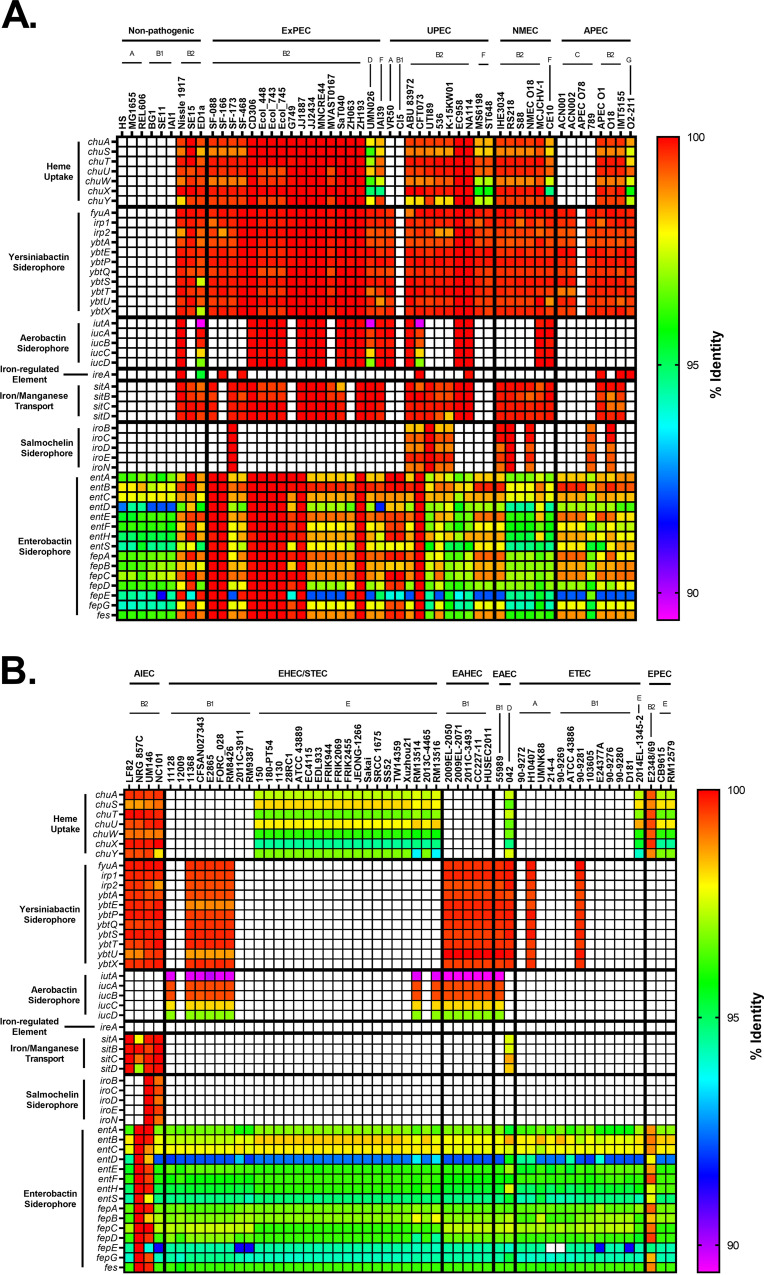
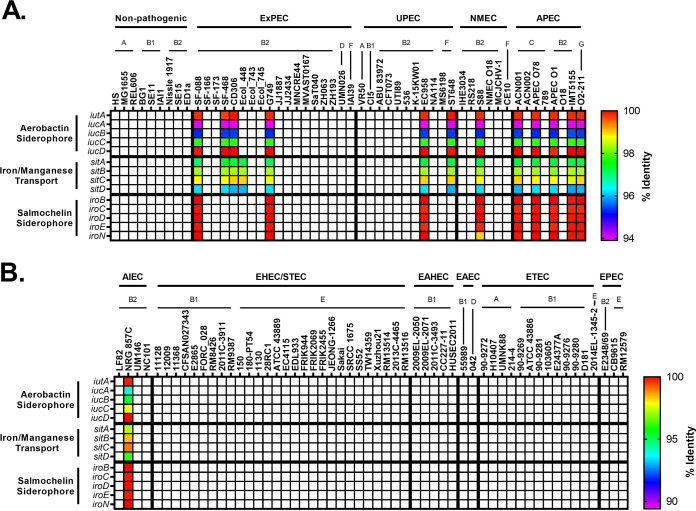
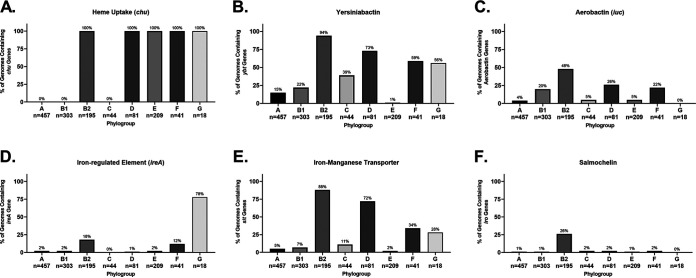
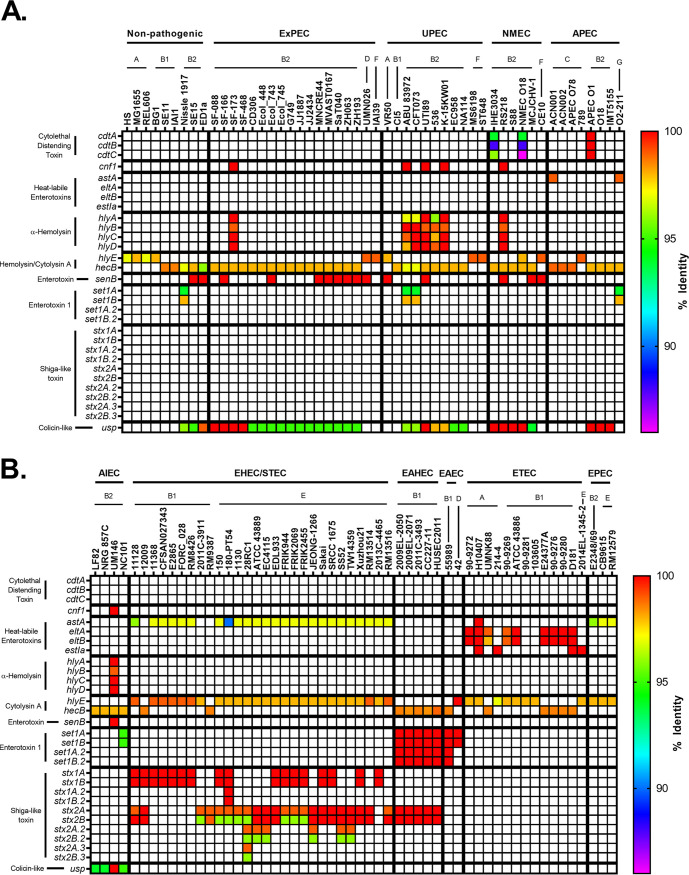
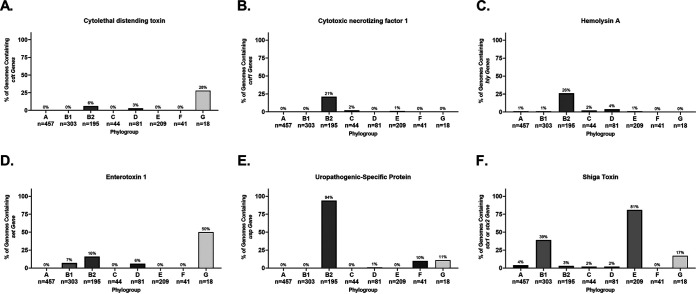
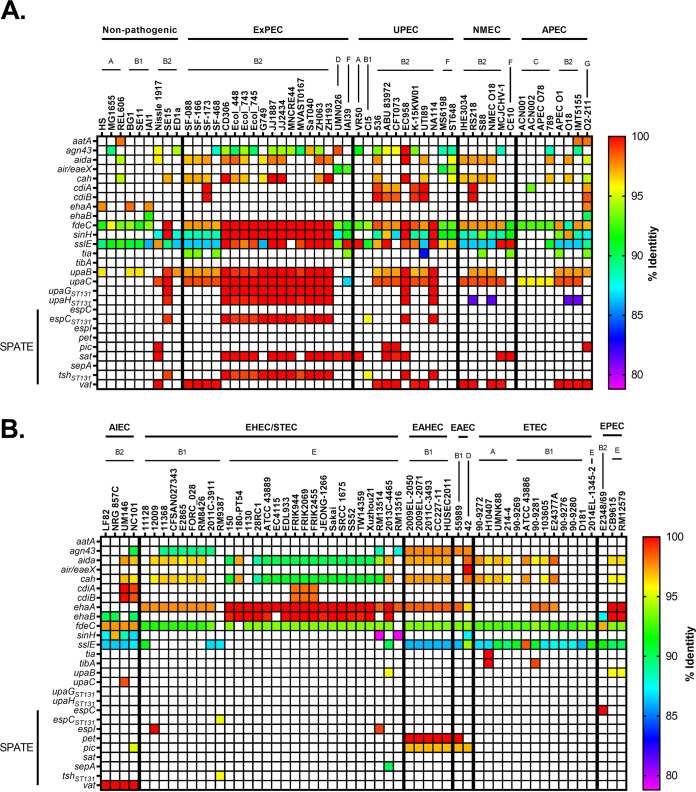
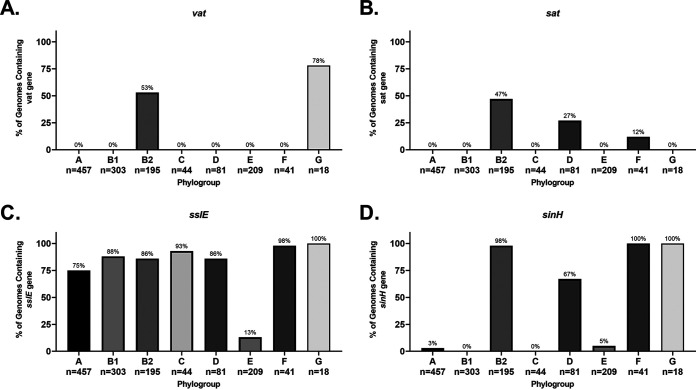
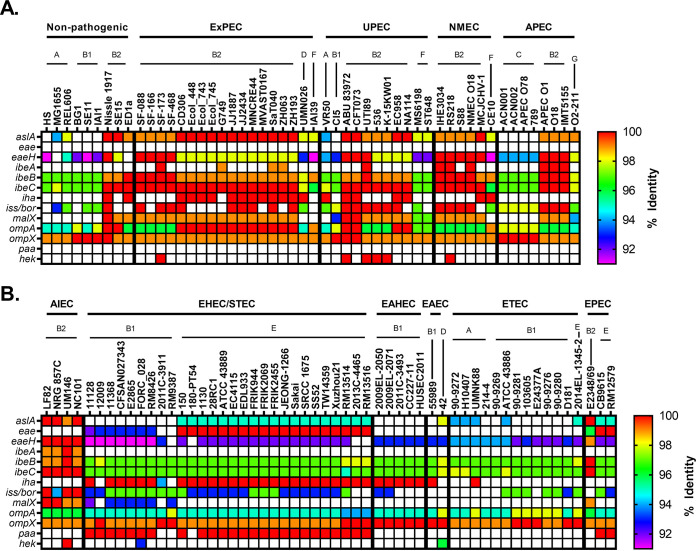
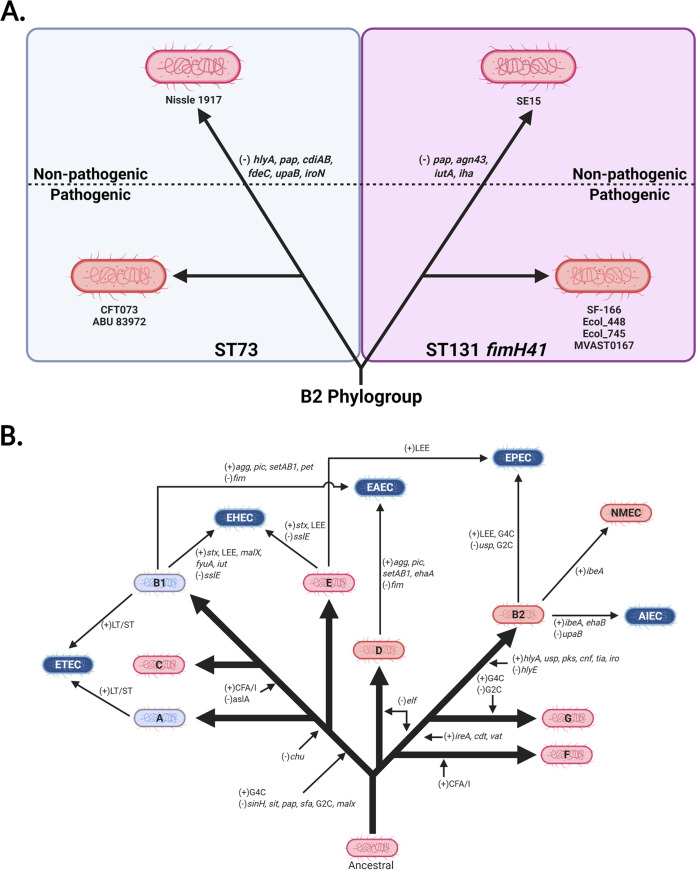
Comparative genomics of bacterial pathogens has been useful for revealing potential virulence factors. Escherichia coli is a significant cause of human morbidity and mortality worldwide but can also exist as a commensal in the human gastrointestinal tract. With many sequenced genomes, it has served as a model organism for comparative genomic studies to understand the link between genetic content and potential for virulence. To date, however, no comprehensive analysis of its complete "virulome" has been performed for the purpose of identifying universal or pathotype-specific targets for vaccine development. Here, we describe the construction of a pathotype database of 107 well-characterized completely sequenced pathogenic and nonpathogenic E. coli strains, which we annotated for major virulence factors (VFs). The data are cross referenced for patterns against pathotype, phylogroup, and sequence type, and the results were verified against all 1,348 complete E. coli chromosomes in the NCBI RefSeq database. Our results demonstrate that phylogroup drives many of the "pathotype-associated" VFs, and ExPEC-associated VFs are found predominantly within the B2/D/F/G phylogenetic clade, suggesting that these phylogroups are better adapted to infect human hosts. Finally, we used this information to propose polyvalent vaccine targets with specificity toward extraintestinal strains, targeting key invasive strategies, including immune evasion (group 2 capsule), iron acquisition (FyuA, IutA, and Sit), adherence (SinH, Afa, Pap, Sfa, and Iha), and toxins (Usp, Sat, Vat, Cdt, Cnf1, and HlyA). While many of these targets have been proposed before, this work is the first to examine their pathotype and phylogroup distribution and how they may be targeted together to prevent disease.
细菌病原体的比较基因组学对于揭示潜在的毒力因子非常有用。大肠杆菌是全世界人类发病率和死亡率的重要原因,但也可以作为人类胃肠道中的共生菌存在。由于有许多测序的基因组,它已成为比较基因组研究的模型生物,以了解遗传物质内容与潜在毒力之间的联系。然而,迄今为止,尚未针对其完整的“毒力组”进行全面分析,以确定用于疫苗开发的通用或病原体特异性靶标。在这里,我们描述了构建一个由 107 种经过充分特征描述的完全测序的致病性和非致病性大肠杆菌菌株组成的病原体数据库,我们对其主要毒力因子(VF)进行了注释。这些数据针对病原体、 phylogroup 和序列类型进行交叉引用,并针对 NCBI RefSeq 数据库中的所有 1348 个完整大肠杆菌染色体进行了验证。我们的结果表明 phylogroup 驱动了许多“与病原体相关”的 VF,而 ExPEC 相关的 VF 主要存在于 B2/D/F/G 进化枝中,这表明这些 phylogroups更适合感染人类宿主。最后,我们利用这些信息提出了针对肠外菌株的多价疫苗靶标,针对关键的侵袭策略,包括免疫逃避(群 2 荚膜)、铁摄取(FyuA、IutA 和 Sit)、粘附(SinH、Afa、Pap、Sfa 和 Iha)和毒素(Usp、Sat、Vat、Cdt、Cnf1 和 HlyA)。虽然许多这些靶标以前已经被提出过,但这项工作是第一个检查它们的病原体和 phylogroup 分布,以及如何将它们一起靶向以预防疾病。