Heidelberg Institute for Theoretical Studies, Heidelberg, Germany.
Interdisciplinary Center for Scientific Computing (IWR), Heidelberg University, Heidelberg, Germany.
PLoS Comput Biol. 2021 May 4;17(5):e1008939. doi: 10.1371/journal.pcbi.1008939. eCollection 2021 May.
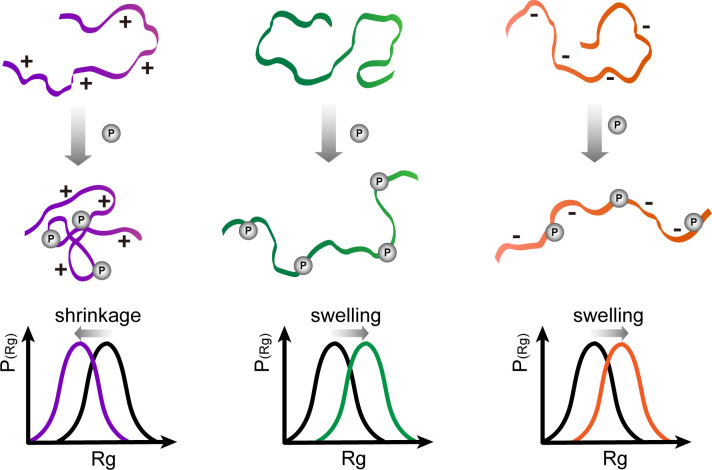
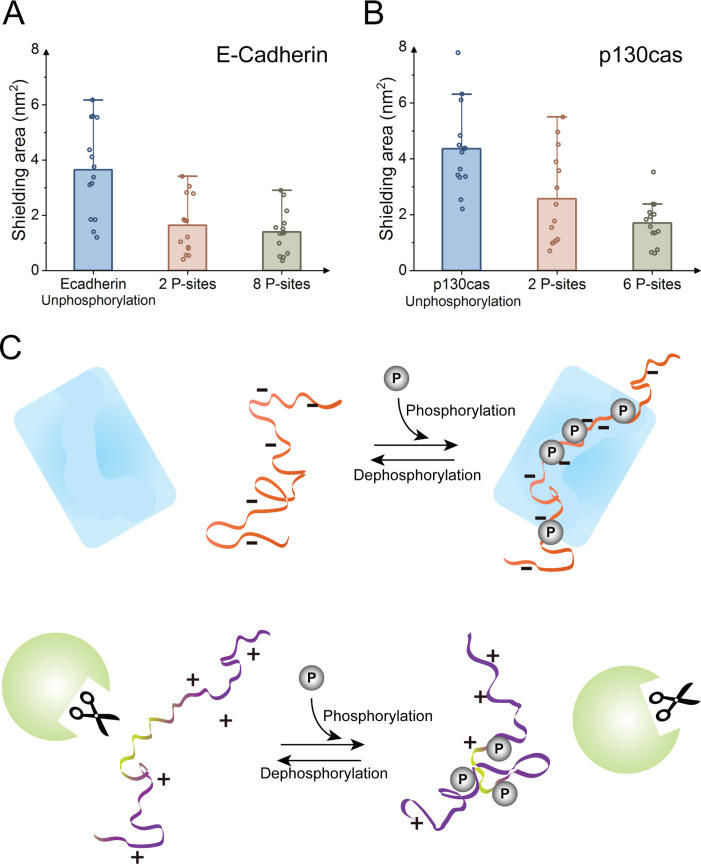
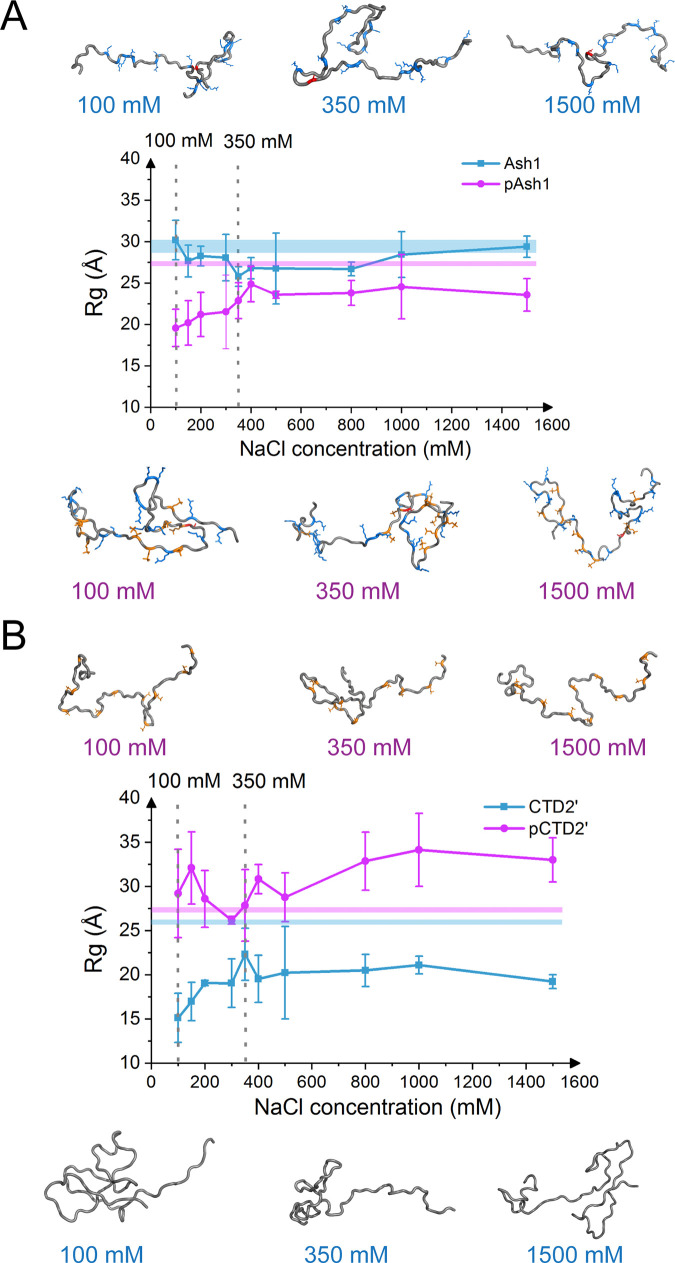
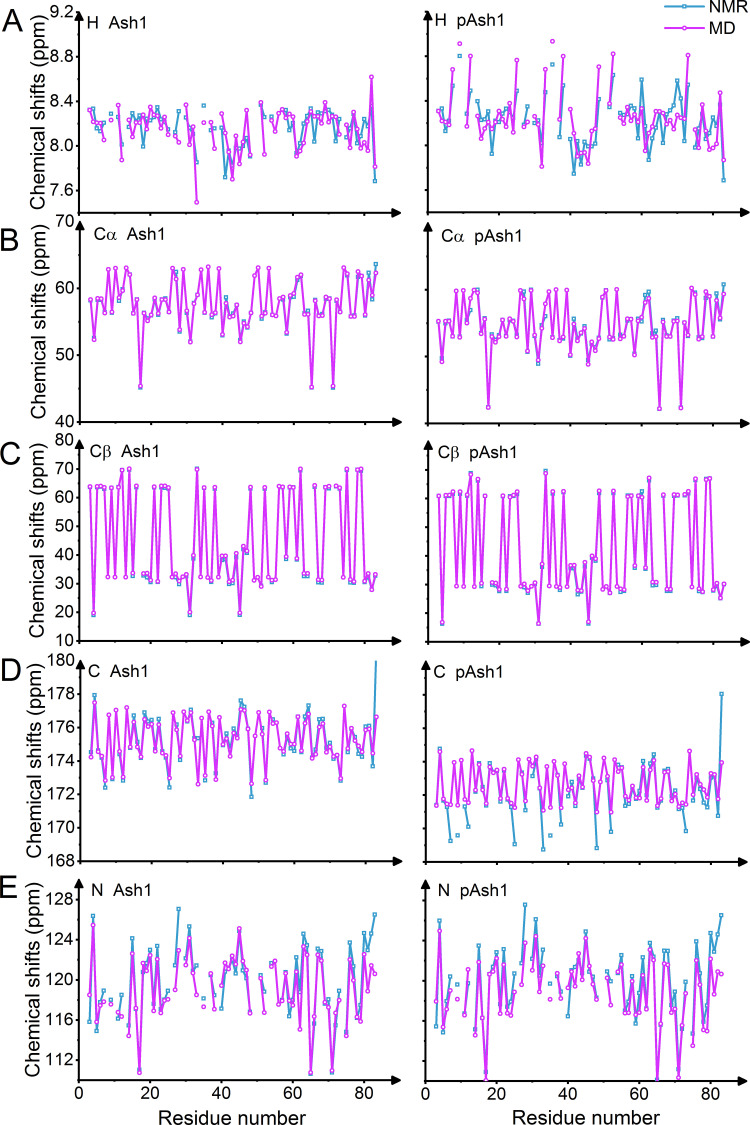
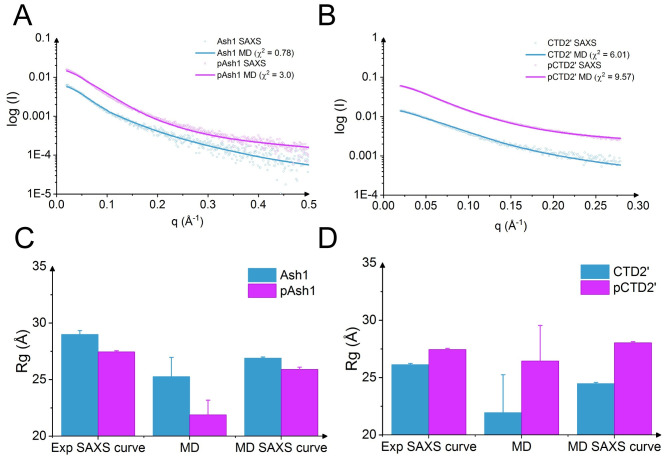
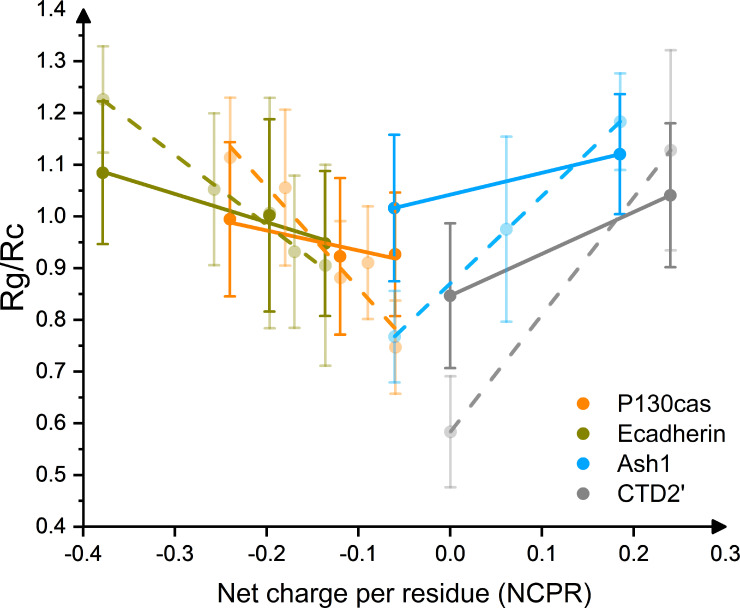
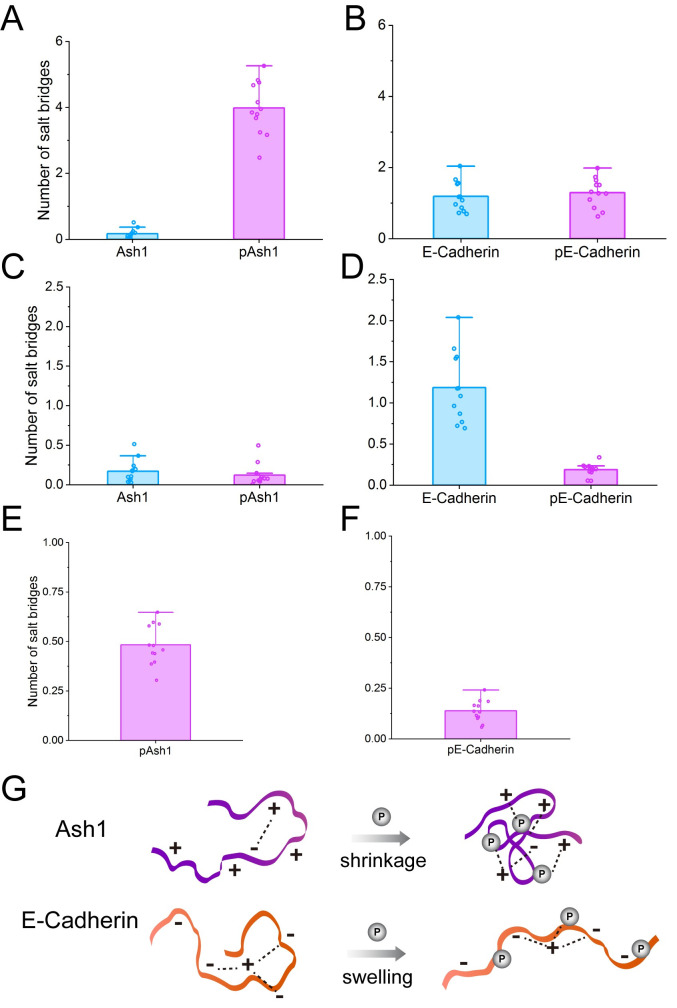
Phosphorylation of intrinsically disordered proteins (IDPs) can produce changes in structural and dynamical properties and thereby mediate critical biological functions. How phosphorylation effects intrinsically disordered proteins has been studied for an increasing number of IDPs, but a systematic understanding is still lacking. Here, we compare the collapse propensity of four disordered proteins, Ash1, the C-terminal domain of RNA polymerase (CTD2'), the cytosolic domain of E-Cadherin, and a fragment of the p130Cas, in unphosphorylated and phosphorylated forms using extensive all-atom molecular dynamics (MD) simulations. We find all proteins to show V-shape changes in their collapse propensity upon multi-site phosphorylation according to their initial net charge: phosphorylation expands neutral or overall negatively charged IDPs and shrinks positively charged IDPs. However, force fields including those tailored towards and commonly used for IDPs overestimate these changes. We find quantitative agreement of MD results with SAXS and NMR data for Ash1 and CTD2' only when attenuating protein electrostatic interactions by using a higher salt concentration (e.g. 350 mM), highlighting the overstabilization of salt bridges in current force fields. We show that phosphorylation of IDPs also has a strong impact on the solvation of the protein, a factor that in addition to the actual collapse or expansion of the IDP should be considered when analyzing SAXS data. Compared to the overall mild change in global IDP dimension, the exposure of active sites can change significantly upon phosphorylation, underlining the large susceptibility of IDP ensembles to regulation through post-translational modifications.
磷酸化的无规则蛋白质(IDPs)可以改变其结构和动力学性质,从而介导关键的生物功能。越来越多的 IDPs 都被研究过磷酸化如何影响无规则蛋白质,但系统的理解仍然缺乏。在这里,我们使用广泛的全原子分子动力学(MD)模拟比较了四种无规则蛋白质的折叠倾向,它们分别是 Ash1、RNA 聚合酶 C 末端结构域(CTD2')、E-钙黏蛋白胞质结构域和 p130Cas 的片段,在非磷酸化和磷酸化形式下。我们发现所有的蛋白质在多位点磷酸化后根据其初始净电荷显示出 V 形变化的折叠倾向:磷酸化扩展了中性或整体带负电荷的 IDPs,并收缩了带正电荷的 IDPs。然而,包括针对和常用于 IDPs 的力场都高估了这些变化。我们发现,只有在使用更高的盐浓度(例如 350mM)来减弱蛋白质静电相互作用时,MD 结果与 Ash1 和 CTD2' 的 SAXS 和 NMR 数据才具有定量一致性,这突出了当前力场中盐桥的过度稳定。我们表明,IDPs 的磷酸化对蛋白质的溶剂化也有很大的影响,这是一个除了 IDP 的实际折叠或扩展之外,在分析 SAXS 数据时还应该考虑的因素。与全局 IDP 尺寸的整体温和变化相比,磷酸化后活性位点的暴露可以显著改变,这强调了 IDP 集合对通过翻译后修饰进行调节的高度敏感性。