Department of Animal Sciences, Wageningen University, Wageningen, The Netherlands.
USDA APHIS National Veterinary Services Laboratories, Ames, Iowa, United States of America.
PLoS One. 2021 May 13;16(5):e0246983. doi: 10.1371/journal.pone.0246983. eCollection 2021.
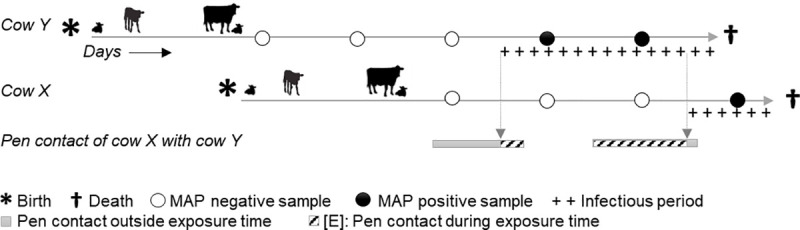
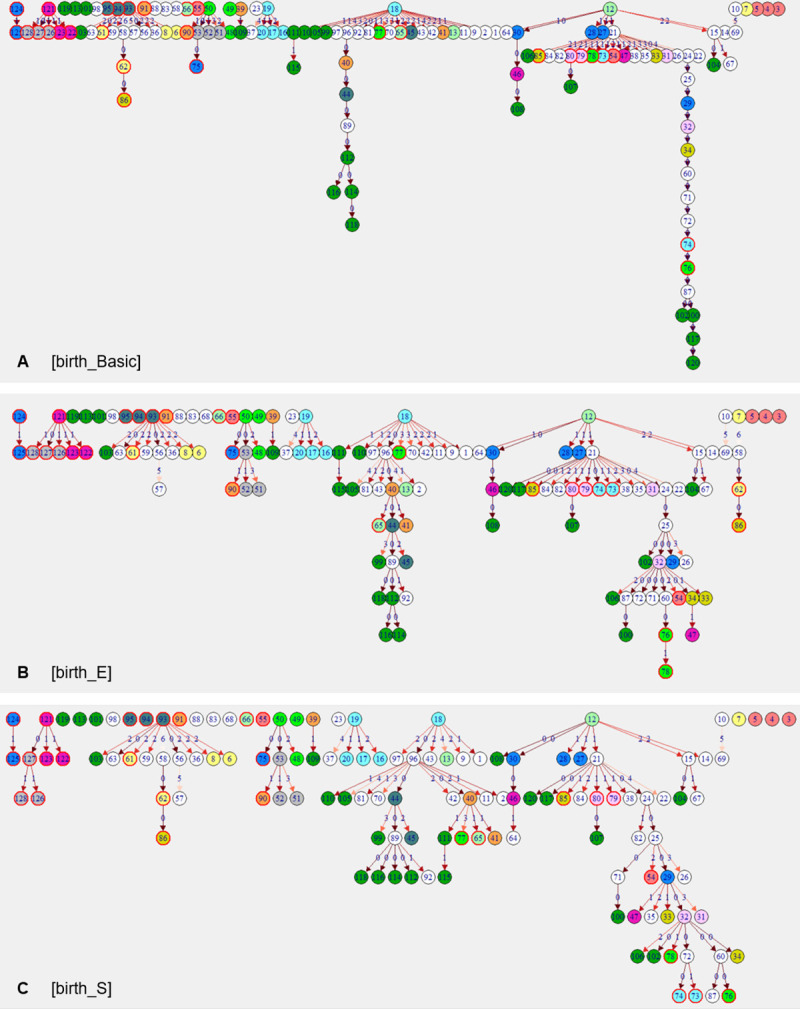
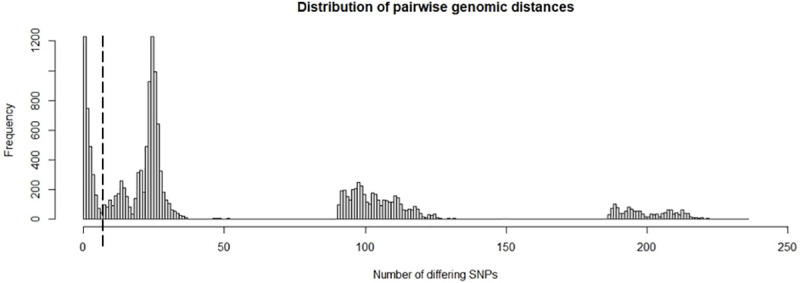
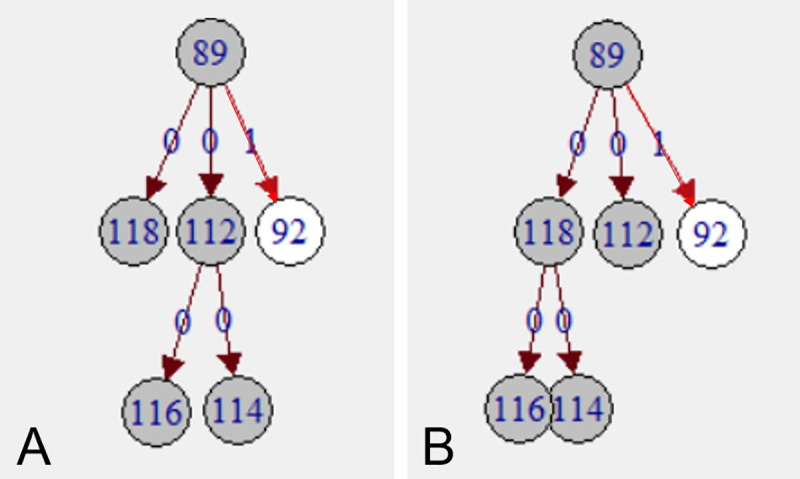
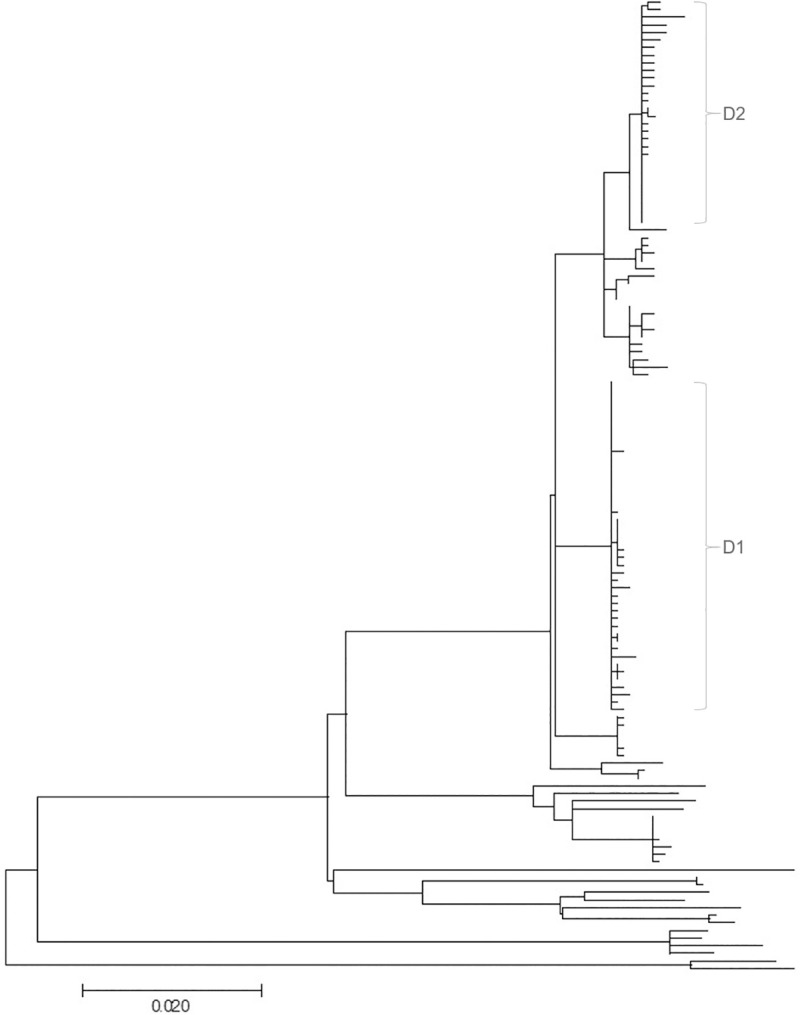
Recent evidence of circulation of multiple strains within herds and mixed infections of cows marks the beginning of a rethink of our knowledge on Mycobacterium avium ssp. paratuberculosis (MAP) epidemiology. Strain typing opens new ways to investigate MAP transmission. This work presents a method for reconstructing infection chains in a setting of endemic Johne's disease on a well-managed dairy farm. By linking genomic data with demographic field data, strain-specific differences in spreading patterns could be quantified for a densely sampled dairy herd. Mixed infections of dairy cows with MAP are common, and some strains spread more successfully. Infected cows remain susceptible for co-infections with other MAP genotypes. The model suggested that cows acquired infection from 1-4 other cows and spread infection to 0-17 individuals. Reconstructed infection chains supported the hypothesis that high shedding animals that started to shed at an early age and showed a progressive infection pattern represented a greater risk for spreading MAP. Transmission of more than one genotype between animals was recorded. In this farm with a good MAP control management program, adult-to-adult contact was proposed as the most important transmission route to explain the reconstructed networks. For each isolate, at least one more likely ancestor could be inferred. Our study results help to capture underlying transmission processes and to understand the challenges of tracing MAP spread within a herd. Only the combination of precise longitudinal field data and bacterial strain type information made it possible to trace infection in such detail.
最近在畜群中循环存在多种菌株以及奶牛的混合感染的证据,标志着我们对分枝杆菌 avium ssp. (MAP)流行病学知识的重新思考的开始。菌株分型为研究 MAP 传播开辟了新的途径。这项工作提出了一种在管理良好的奶牛场中对地方性约翰氏病进行设置的情况下重建感染链的方法。通过将基因组数据与人口统计学现场数据相关联,可以量化在密集采样的奶牛群中菌株特异性传播模式的差异。奶牛的 MAP 混合感染很常见,有些菌株传播得更成功。受感染的奶牛仍然容易受到其他 MAP 基因型的再感染。该模型表明,奶牛从 1 到 4 头其他奶牛感染,并传播感染至 0 到 17 头个体。重建的感染链支持了这样的假设,即早期开始排菌且具有进行性感染模式的高排菌动物代表着传播 MAP 的更大风险。记录了动物之间传播一种以上基因型。在这个具有良好 MAP 控制管理计划的农场中,建议成年到成年的接触是解释重建网络的最重要传播途径。对于每个分离株,至少可以推断出一个更可能的祖先。我们的研究结果有助于捕捉潜在的传播过程,并了解在牛群中追踪 MAP 传播的挑战。只有精确的纵向现场数据和细菌菌株类型信息的组合才有可能如此详细地追踪感染。