Saxenborn Patricia, Baxter John, Tilevik Andreas, Fagerlind Magnus, Dyrkell Fredrik, Pernestig Anna-Karin, Enroth Helena, Tilevik Diana
Systems Biology Research Centre, School of Bioscience, University of Skövde, Skövde, Sweden.
1928 Diagnostics, Gothenburg, Sweden.
Front Microbiol. 2021 Apr 30;12:640408. doi: 10.3389/fmicb.2021.640408. eCollection 2021.
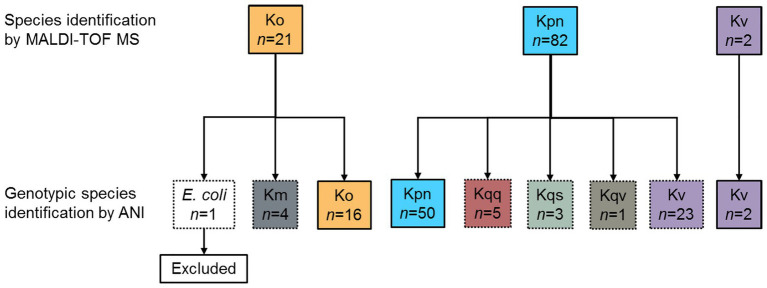
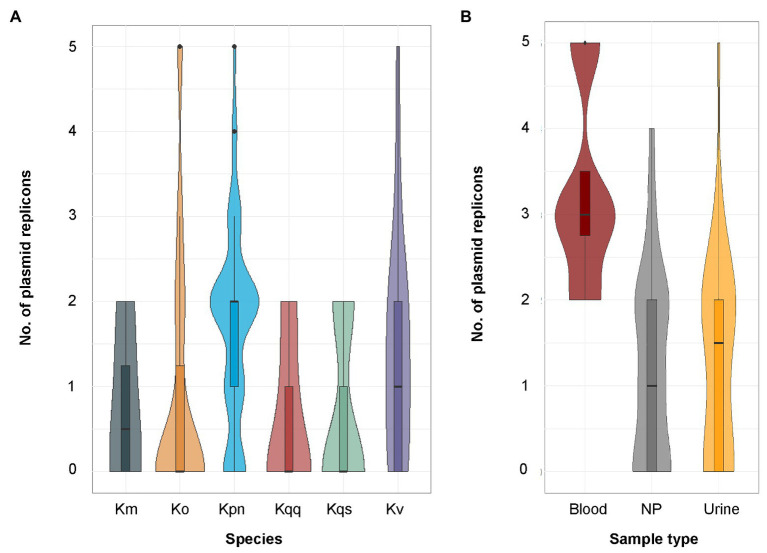
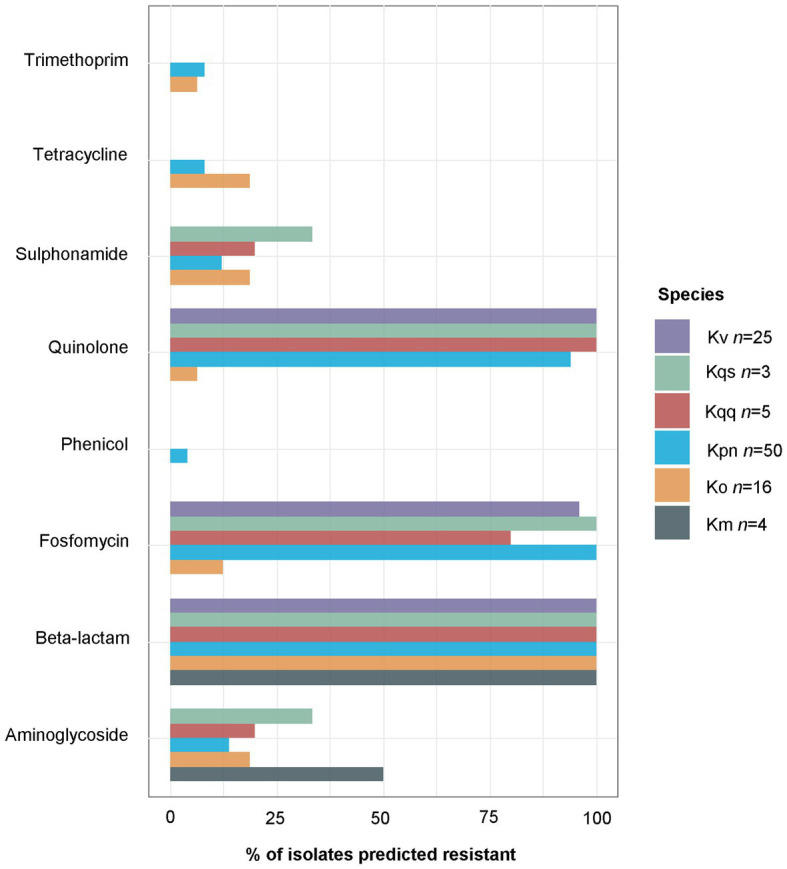
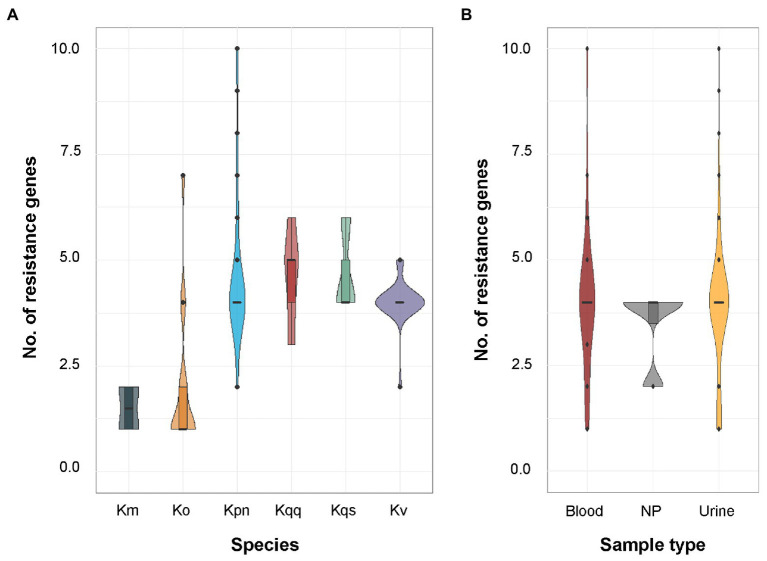
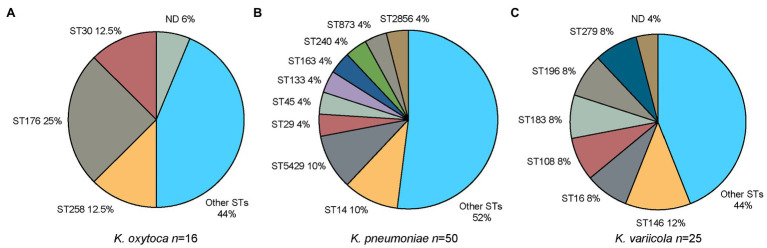
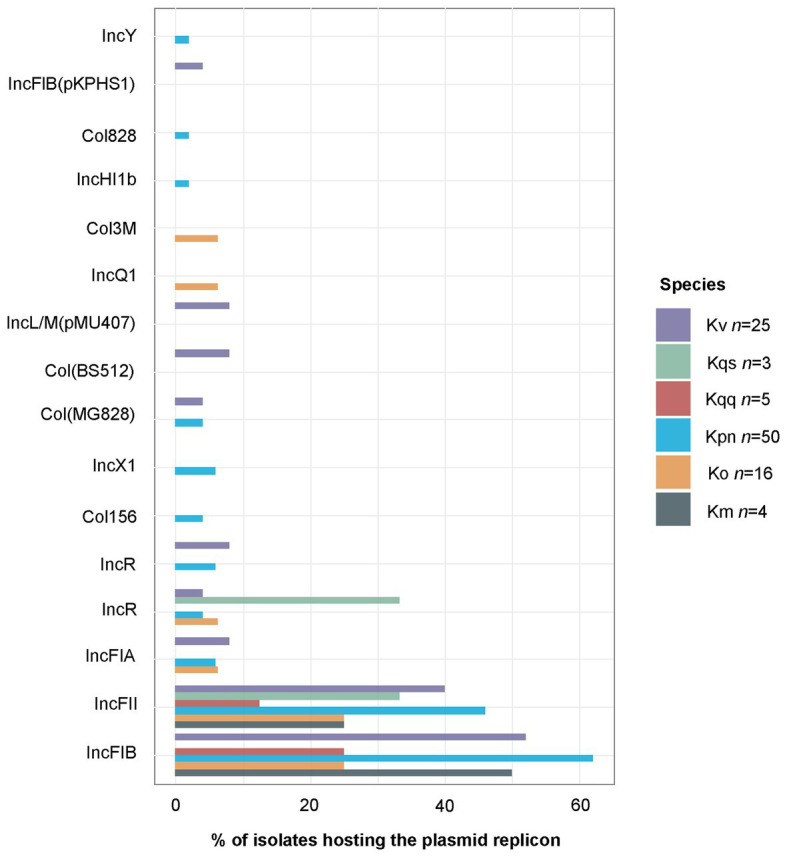
is a genus of Gram-negative bacteria known to be opportunistic pathogens that may cause a variety of infections in humans. Highly drug-resistant species, especially , have emerged rapidly and are becoming a major concern in clinical management. Although is considered the most important pathogen within the genus, the true clinical significance of the other species is likely underrecognized due to the inability of conventional microbiological methods to distinguish between the species leading to high rates of misidentification. Bacterial whole-genome sequencing (WGS) enables precise species identification and characterization that other technologies do not allow. Herein, we have characterized the diversity and traits of spp. in community-onset infections by WGS of clinical isolates ( = 105) collected during a prospective sepsis study in Sweden. The sequencing revealed that 32 of the 82 isolates (39.0%) initially identified as with routine microbiological methods based on cultures followed by matrix-assisted laser desorption-time of flight mass spectrometry (MALDI-TOF MS) had been misidentified. Of these, 23 were identified as and nine as other members of the complex. Comparisons of the number of resistance genes showed that significantly fewer resistance genes were detected in compared to and (both values of < 0.001). Moreover, a high proportion of the isolates within the complex were predicted to be genotypically multidrug-resistant (MDR; 79/84, 94.0%) in contrast to (3/16, 18.8%) and (0/4, 0.0%). All isolates predicted as genotypically MDR were found to harbor the combination of β-lactam, fosfomycin, and quinolone resistance markers. Multi-locus sequence typing (MLST) revealed a high diversity of sequence types among the spp. with ST14 (10.0%) and ST5429 (10.0%) as the most prevalent ones for , ST146 for (12.0%), and ST176 for (25.0%). In conclusion, the results from this study highlight the importance of using high-resolution genotypic methods for identification and characterization of clinical spp. isolates. Our findings indicate that infections caused by other members of the complex than are a more common clinical problem than previously described, mainly due to high rates of misidentifications.
是革兰氏阴性菌的一个属,已知为机会性病原体,可在人类中引起多种感染。高耐药性物种,尤其是,迅速出现,正成为临床管理中的一个主要问题。尽管被认为是该属中最重要的病原体,但由于传统微生物学方法无法区分这些物种,导致错误鉴定率很高,其他物种的真正临床意义可能未得到充分认识。细菌全基因组测序(WGS)能够实现精确的物种鉴定和特征描述,这是其他技术所不具备的。在此,我们通过对瑞典一项前瞻性败血症研究中收集的临床分离株(=105)进行WGS,对社区获得性感染中的 spp. 的多样性和特征进行了描述。测序结果显示,在最初通过基于培养的常规微生物学方法,随后用基质辅助激光解吸飞行时间质谱(MALDI-TOF MS)鉴定为的82株分离株中,有32株(39.0%)被错误鉴定。其中,23株被鉴定为,9株被鉴定为复合体的其他成员。对耐药基因数量的比较表明,与和相比,中检测到的耐药基因明显较少(两者值均<0.001)。此外,复合体中的分离株中有很大比例预计为基因型多重耐药(MDR;79/84,94.0%),而相比之下,为(3/16,18.8%),为(0/4,0.0%)。所有预测为基因型MDR的分离株均被发现携带β-内酰胺、磷霉素和喹诺酮耐药标记的组合。多位点序列分型(MLST)显示,spp. 中的序列类型具有高度多样性,其中ST14(10.0%)和ST5429(10.0%)是最常见的,为ST146(12.0%),为ST176(25.0%)。总之,本研究结果突出了使用高分辨率基因型方法对临床spp. 分离株进行鉴定和特征描述的重要性。我们的研究结果表明,由复合体中除之外的其他成员引起的感染是一个比以前描述的更常见的临床问题,主要是由于错误鉴定率很高。