Department of Pediatrics, Military Teaching Hospital Mohammed V, Faculty of Medicine and Pharmacy, University Mohammed V, Rabat, Morocco.
Department of Pediatrics, Children's Hospital, Faculty of Medicine and Pharmacy, University Mohammed V, Rabat, Morocco.
Pan Afr Med J. 2021 Feb 18;38:188. doi: 10.11604/pamj.2021.38.188.27941. eCollection 2021.
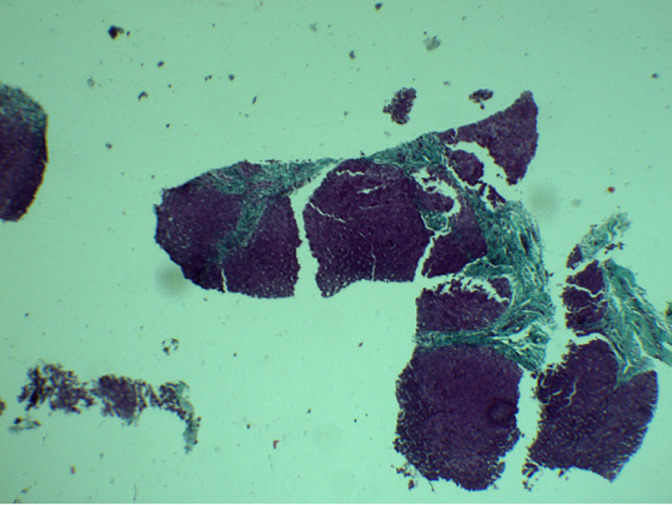
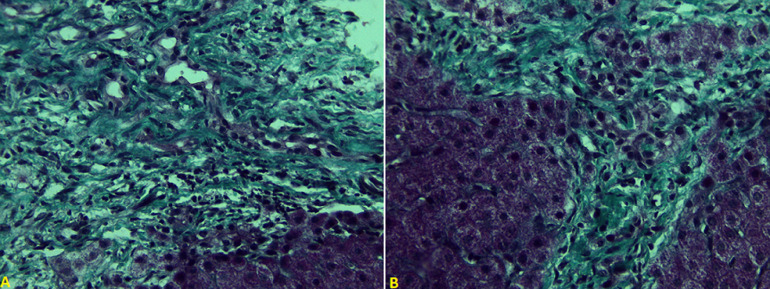
Congenital hepatic fibrosis (CHF) is a rare autosomal recessive disease derived from biliary dysgenesis secondary to ductal plate malformation; it often coexists with Caroli's disease, von Meyenburg complexes, autosomal dominant polycystic kidney disease (ADPKD), and autosomal recessive polycystic kidney disease (ARPKD). Although CHF was first named and described in detail by Kerr et al. in 1961. Its pathogenesis still remains unclear. The exact incidence and prevalence are not known, and only a few hundred patients with CHF have been reported in the literature to date. However, with the development of noninvasive diagnostic techniques such as ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI), CHF may now be more frequently detected. Anatomopathological examination of liver biopsy is the gold standard in diagnosis of CHF. Patients with CHF exhibit variable clinical presentations, ranging from no symptoms to severe symptoms such as acute hepatic decompensation and even cirrhosis. The most common presentations in these patients are splenomegaly, esophageal varices, and gastrointestinal bleeding due to portal hypertension. In addition, in younger children, CHF often is accompanied by renal cysts or increased renal echogenicity. Great variability exists among the signs and symptoms of the disease from early childhood to the 5 or 6 decade of life, and in most patients the disorder is diagnosed during adolescence or young adulthood. Here, we present two cases of congenital hepatic fibrosis in 2-years-old girl and 12-year-old male who had been referred for evaluation of an abdominal distension with persistent hyper-transaminasemia and cholestasis, the diagnostic was made according to the results of medical imaging (CT or MRI), a liver biopsy, and genetic testing.
先天性肝纤维化(CHF)是一种罕见的常染色体隐性疾病,源自胆管发育不良继发于胆管板畸形;它常与 Caroli 病、von Meyenburg 复合体、常染色体显性多囊肾病(ADPKD)和常染色体隐性多囊肾病(ARPKD)共存。尽管 CHF 于 1961 年由 Kerr 等人首次命名并详细描述,但其发病机制仍不清楚。确切的发病率和患病率尚不清楚,迄今为止,文献中仅报道了几百例 CHF 患者。然而,随着超声、计算机断层扫描(CT)和磁共振成像(MRI)等非侵入性诊断技术的发展,CHF 现在可能更频繁地被发现。肝活检的解剖病理学检查是 CHF 诊断的金标准。CHF 患者表现出不同的临床表现,从无症状到急性肝功能失代偿甚至肝硬化等严重症状不等。这些患者最常见的表现是脾肿大、食管静脉曲张和门脉高压引起的胃肠道出血。此外,在年幼的儿童中,CHF 常伴有肾囊肿或肾脏回声增强。从儿童早期到 50 或 60 岁,疾病的体征和症状存在很大差异,在大多数患者中,该疾病在青少年或成年早期被诊断出来。在这里,我们介绍了 2 例 2 岁女孩和 12 岁男性的先天性肝纤维化病例,他们因持续性高转氨酶血症和胆汁淤积引起的腹部膨隆而被转诊评估,根据医学影像学(CT 或 MRI)、肝活检和基因检测结果做出了诊断。