Department of Anesthesiology, Heidelberg University Hospital, Heidelberg, Germany.
PLoS One. 2021 May 17;16(5):e0251800. doi: 10.1371/journal.pone.0251800. eCollection 2021.
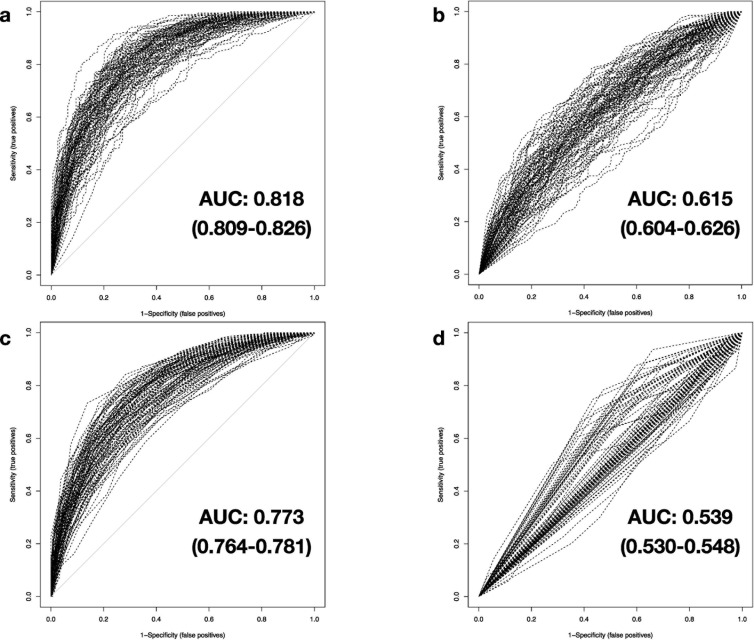
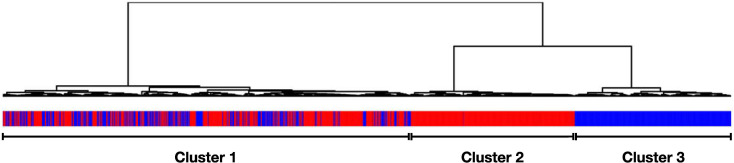
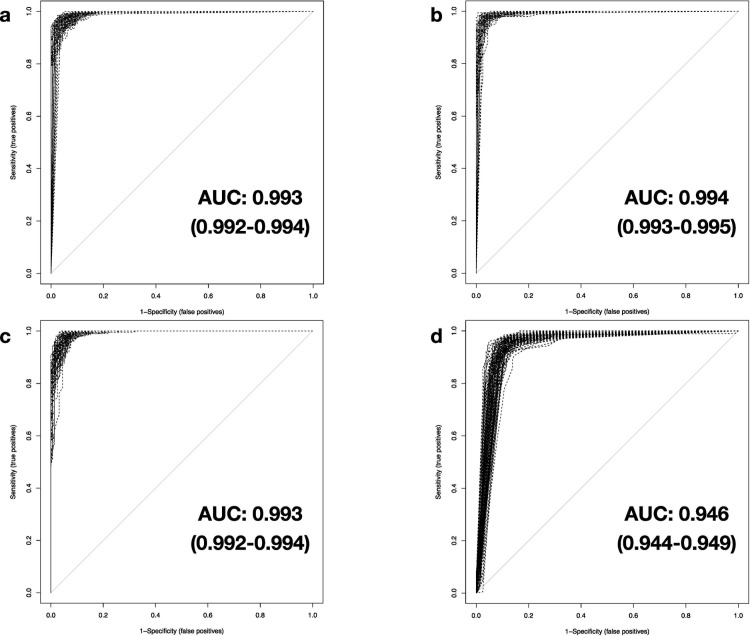
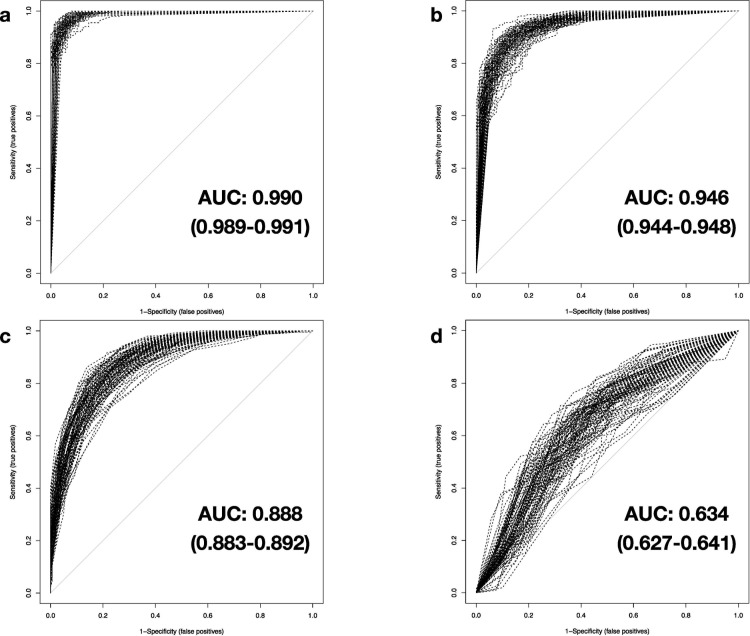
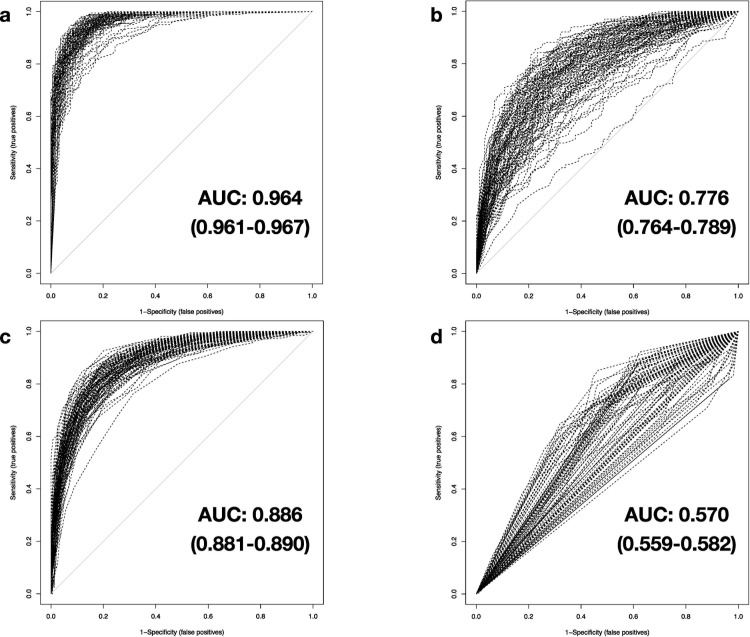
We investigate the feasibility of molecular-level sample classification of sepsis using microarray gene expression data merged by in silico meta-analysis. Publicly available data series were extracted from NCBI Gene Expression Omnibus and EMBL-EBI ArrayExpress to create a comprehensive meta-analysis microarray expression set (meta-expression set). Measurements had to be obtained via microarray-technique from whole blood samples of adult or pediatric patients with sepsis diagnosed based on international consensus definition immediately after admission to the intensive care unit. We aggregate trauma patients, systemic inflammatory response syndrome (SIRS) patients, and healthy controls in a non-septic entity. Differential expression (DE) analysis is compared with machine-learning-based solutions like decision tree (DT), random forest (RF), support vector machine (SVM), and deep-learning neural networks (DNNs). We evaluated classifier training and discrimination performance in 100 independent iterations. To test diagnostic resilience, we gradually degraded expression data in multiple levels. Clustering of expression values based on DE genes results in partial identification of sepsis samples. In contrast, RF, SVM, and DNN provide excellent diagnostic performance measured in terms of accuracy and area under the curve (>0.96 and >0.99, respectively). We prove DNNs as the most resilient methodology, virtually unaffected by targeted removal of DE genes. By surpassing most other published solutions, the presented approach substantially augments current diagnostic capability in intensive care medicine.
我们研究了通过计算机元分析合并的微阵列基因表达数据对脓毒症进行分子水平样本分类的可行性。从 NCBI Gene Expression Omnibus 和 EMBL-EBI ArrayExpress 提取公开可用的数据集,创建一个综合的元分析微阵列表达集(元表达集)。测量必须通过微阵列技术从在重症监护病房入院后立即根据国际共识定义诊断为脓毒症的成年或儿科患者的全血样本中获得。我们将创伤患者、全身性炎症反应综合征(SIRS)患者和健康对照聚集在一个非脓毒症实体中。差异表达(DE)分析与基于机器学习的解决方案(如决策树(DT)、随机森林(RF)、支持向量机(SVM)和深度学习神经网络(DNN))进行比较。我们在 100 次独立迭代中评估了分类器的训练和区分性能。为了测试诊断的弹性,我们在多个级别逐渐降低表达数据。基于 DE 基因的表达值聚类导致部分识别脓毒症样本。相比之下,RF、SVM 和 DNN 以准确性和曲线下面积(分别>0.96 和>0.99)衡量,提供了出色的诊断性能。我们证明 DNN 是最具弹性的方法,几乎不受 DE 基因的靶向去除的影响。通过超越大多数其他已发表的解决方案,所提出的方法大大增强了重症监护医学中的当前诊断能力。