Department of Genome Sciences, University of Washington, Seattle, WA 98195;
Computational Biology Program, Fred Hutchinson Cancer Research Center, Seattle, WA 98109.
Proc Natl Acad Sci U S A. 2021 May 25;118(21). doi: 10.1073/pnas.2013798118.
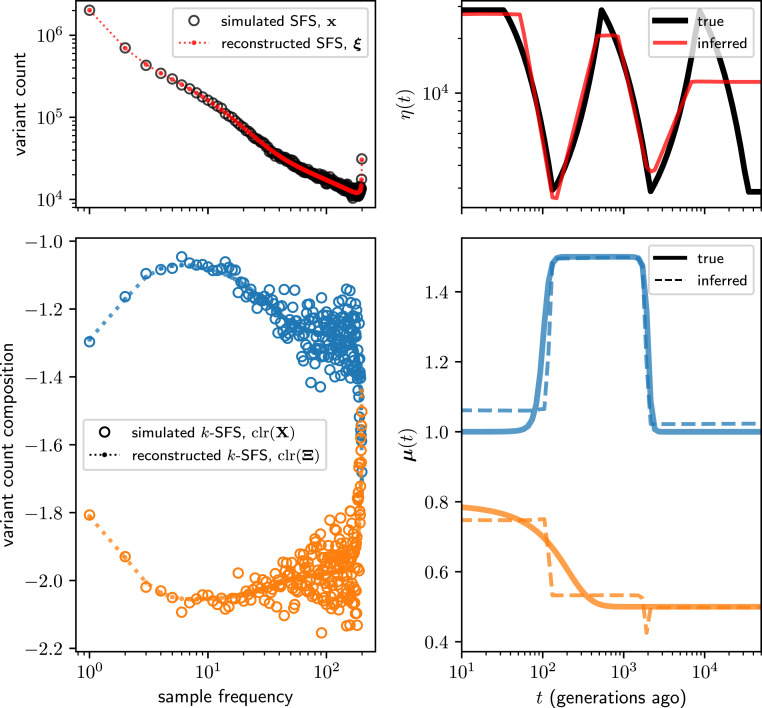
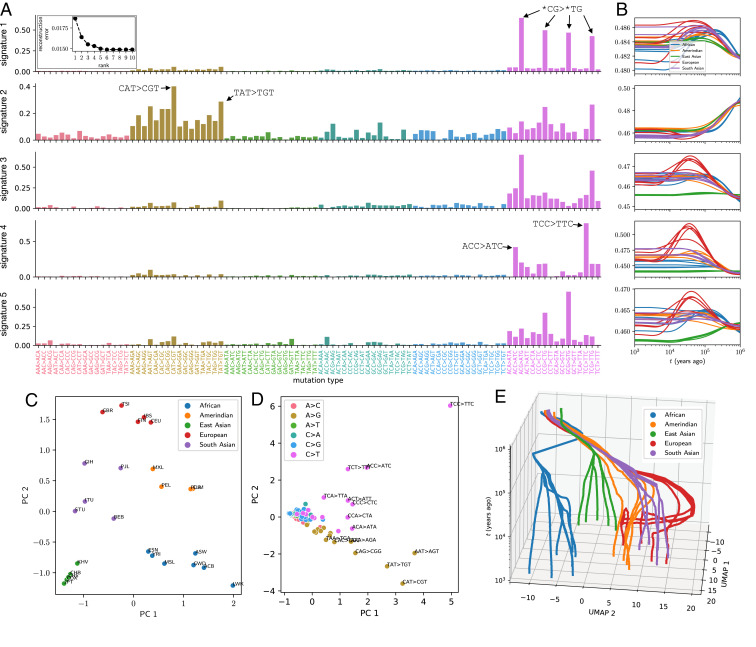
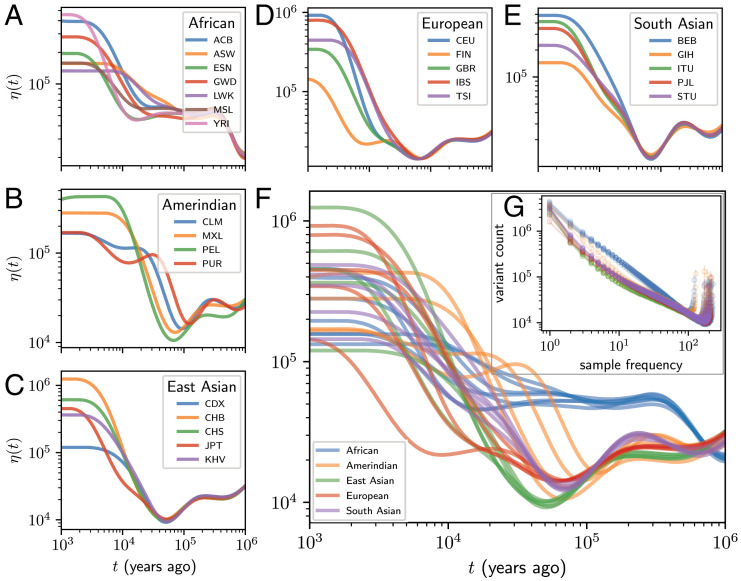
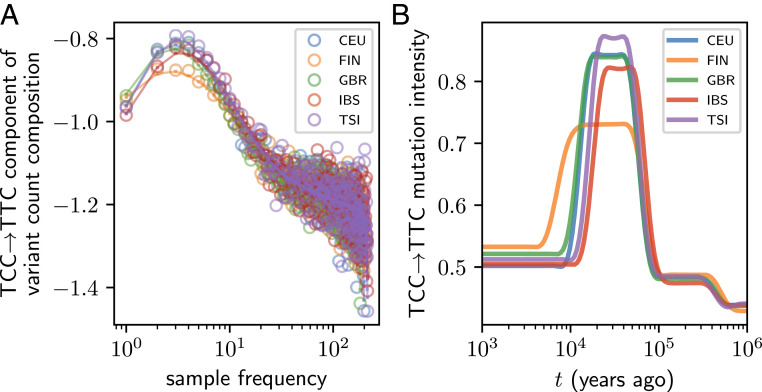
As populations boom and bust, the accumulation of genetic diversity is modulated, encoding histories of living populations in present-day variation. Many methods exist to decode these histories, and all must make strong model assumptions. It is typical to assume that mutations accumulate uniformly across the genome at a constant rate that does not vary between closely related populations. However, recent work shows that mutational processes in human and great ape populations vary across genomic regions and evolve over time. This perturbs the mutation spectrum (relative mutation rates in different local nucleotide contexts). Here, we develop theoretical tools in the framework of Kingman's coalescent to accommodate mutation spectrum dynamics. We present mutation spectrum history inference (mushi), a method to perform nonparametric inference of demographic and mutation spectrum histories from allele frequency data. We use mushi to reconstruct trajectories of effective population size and mutation spectrum divergence between human populations, identify mutation signatures and their dynamics in different human populations, and calibrate the timing of a previously reported mutational pulse in the ancestors of Europeans. We show that mutation spectrum histories can be placed in a well-studied theoretical setting and rigorously inferred from genomic variation data, like other features of evolutionary history.
随着人口的兴衰,遗传多样性的积累被调节,将现存人口的历史编码在当今的变化中。有许多方法可以解码这些历史,所有这些方法都必须做出强有力的模型假设。通常假设突变在基因组中以恒定的速率均匀积累,而在密切相关的种群之间不会发生变化。然而,最近的研究表明,人类和大型猿类种群中的突变过程在基因组区域之间存在差异,并随时间演变。这扰乱了突变谱(不同局部核苷酸环境中的相对突变率)。在这里,我们在 Kingman 的合并框架内开发了理论工具来适应突变谱动力学。我们提出了突变谱历史推断(mushi),这是一种从等位基因频率数据中进行人口和突变谱历史的非参数推断的方法。我们使用 mushi 来重建人类群体之间有效种群大小和突变谱分歧的轨迹,识别不同人类群体中的突变特征及其动态,并校准欧洲人祖先中先前报道的突变脉冲的时间。我们表明,突变谱历史可以置于一个经过充分研究的理论框架中,并从基因组变异数据中进行严格推断,就像进化历史的其他特征一样。