JetBrains Research Department, Space Office Center. Address: Primorskiy pr. 70, building 1, 197374, St.Petersburg, Russia.
Department of Pathology and Immunology, 660 S Euclid Ave, St. Louis, MO 63110, USA.
Bioinformatics. 2021 Nov 18;37(22):4235-4237. doi: 10.1093/bioinformatics/btab376.
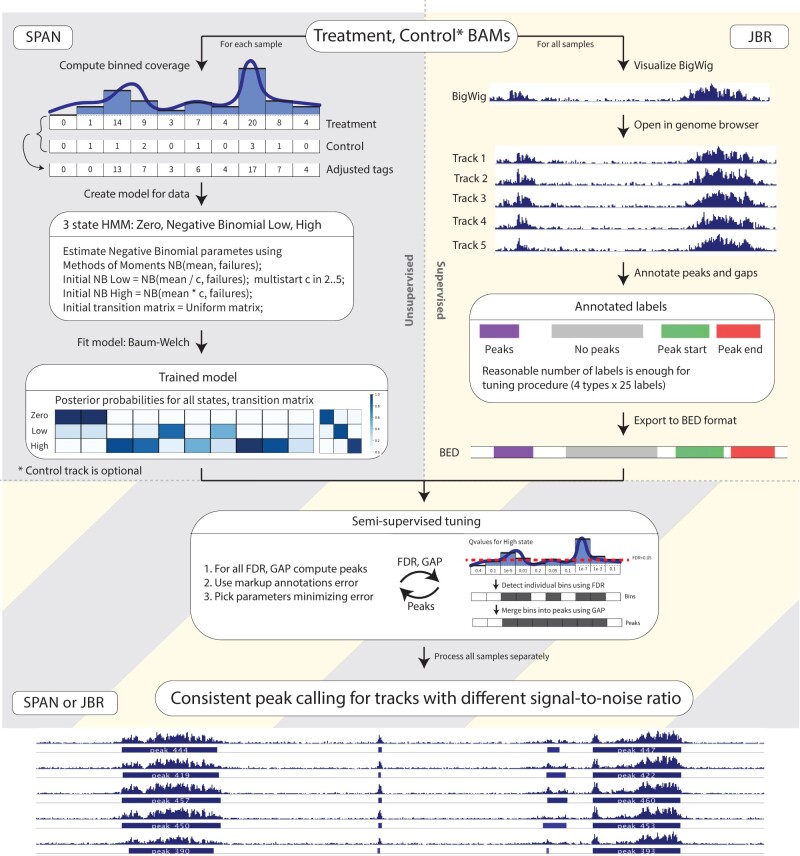
The widespread application of ChIP-seq led to a growing need for consistent analysis of multiple epigenetics profiles, for instance, in human studies where multiple replicates are a common element of design. Such multi-samples experimental designs introduced analytical and computational challenges. For example, when peak calling is done independently for each sample, small differences in signal strength/quality lead to a very different number of peaks for individual samples, making group-level analysis difficult. On the other side, when samples are pooled together for joint analysis, individual-level statistical differences are averaged out. Recently, we have demonstrated that a semi-supervised peak calling approach (SPAN) allows for robust analysis of multiple epigenetic profiles while preserving individual sample statistics. Here, we present this approach's implementation, centered around the JBR genome browser, a stand-alone tool that allows for accessible and streamlined annotation, analysis and visualization. Specifically, JBR supports graphical interactive manual region selection and annotation, thereby addressing supervised learning's key procedural challenge. Furthermore, JBR includes the capability for peak optimization, i.e. calibration of sample-specific peak calling parameters by leveraging manual annotation. This procedure can be applied to a broad range of ChIP-seq datasets of different quality and chromatin accessibility ATAC-seq, including single-cell experiments. JBR was designed for efficient data processing, resulting in fast viewing and analysis of multiple replicates, up to thousands of tracks. Accelerated execution and integrated semi-supervised peak calling make JBR and SPAN next-generation visualization and analysis tools for multi-sample epigenetic data.
SPAN and JBR run on Linux, Mac OS and Windows, and is freely available at https://research.jetbrains.org/groups/biolabs/tools/span-peak-analyzer and https://research.jetbrains.org/groups/biolabs/tools/jbr-genome-browser.
Supplementary data are available at Bioinformatics online.
ChIP-seq 的广泛应用导致了对多个表观遗传学谱进行一致分析的需求不断增长,例如在人类研究中,多个重复是设计的常见元素。这种多样本实验设计带来了分析和计算方面的挑战。例如,当针对每个样本独立进行峰调用时,信号强度/质量的微小差异会导致各个样本的峰数量非常不同,从而使组水平分析变得困难。另一方面,当将样本合并在一起进行联合分析时,个体水平的统计差异会被平均化。最近,我们已经证明,一种半监督峰调用方法(SPAN)允许在保留个体样本统计信息的同时,对多个表观遗传学谱进行稳健分析。在这里,我们介绍了这种方法的实现,它以 JBR 基因组浏览器为中心,这是一个独立的工具,允许进行可访问和简化的注释、分析和可视化。具体来说,JBR 支持图形化交互手动区域选择和注释,从而解决了监督学习的关键程序挑战。此外,JBR 还包括峰优化的功能,即通过利用手动注释来校准样本特定的峰调用参数。此过程可应用于不同质量和染色质可及性 ATAC-seq 的广泛的 ChIP-seq 数据集,包括单细胞实验。JBR 旨在实现高效的数据处理,从而实现对多个重复的快速查看和分析,最多可达数千个轨道。加速执行和集成的半监督峰调用使 JBR 和 SPAN 成为用于多样本表观遗传学数据的下一代可视化和分析工具。
SPAN 和 JBR 可在 Linux、Mac OS 和 Windows 上运行,可在 https://research.jetbrains.org/groups/biolabs/tools/span-peak-analyzer 和 https://research.jetbrains.org/groups/biolabs/tools/jbr-genome-browser 上免费获得。
补充数据可在 Bioinformatics 在线获得。