Chen Cheng, Fu Rongtao, Wang Jian, Li Xingyue, Chen Xiaojuan, Li Qiang, Lu Daihua
Institute of Plant Protection, Sichuan Academy of Agricultural Sciences, Key Laboratory of Integrated Pest Management on Crops in Southwest, Ministry of Agriculture, Chengdu, PR China.
Key Laboratory of Coarse Cereal Processing, Ministry of Agriculture and Rural Affairs, School of Food and Biological Engineering, Chengdu University, Chengdu, PR China.
Comput Struct Biotechnol J. 2021 Apr 27;19:2607-2617. doi: 10.1016/j.csbj.2021.04.065. eCollection 2021.
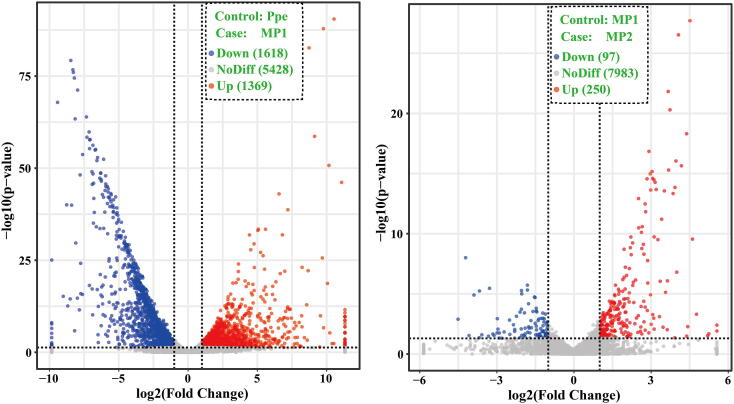
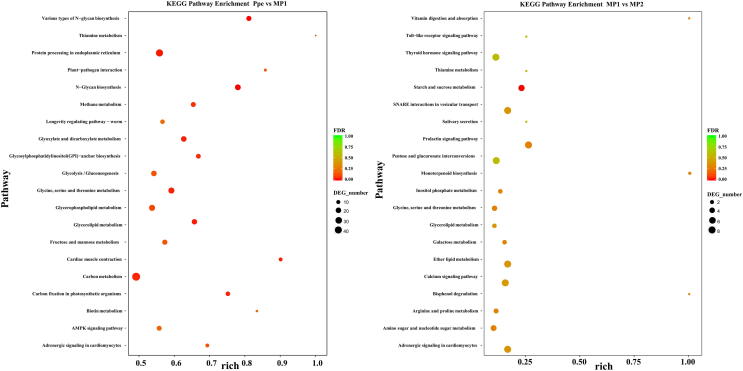
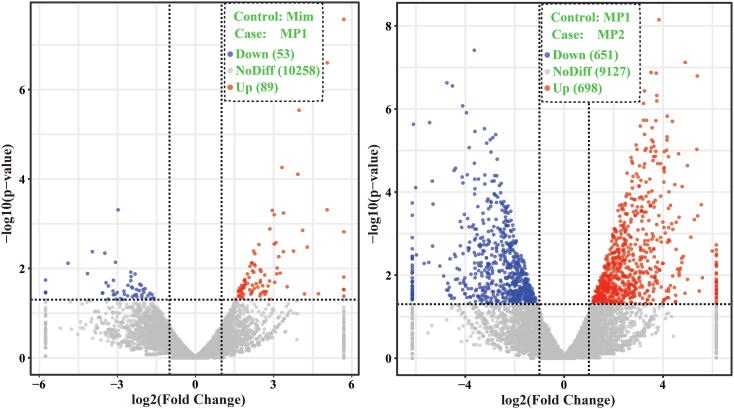
is one of the pathogens of morels, which greatly affects the yield and quality of spp.. In the present study, we assembled the genome sequence of the fungus SAAS_ppe1. We analyzed the transcriptional profile of SAAS_ppe1 infection of at different stages (3 days and 6 days after infection) and the response of using the transcriptome. The assembled genome sequence of SAAS_ppe1 was 39.78 Mb in length (11 scaffolds; scaffold N50, 6.50 Mb), in which 99.7% of the expected genes were detected. A total of 7.48% and 19.83% clean transcriptional reads from the infected sites were mapped to the genome at the early and late stages of infection, respectively. There were 3,943 genes differently expressed in at different stages of infection, of which 24 genes had increased expression with the infection and infection stage, including diphthamide biosynthesis, aldehyde reductase, and NAD (P)H-hydrate epimerase ( < 0.05). Several genes had variable expression trends at different stages of infection, indicating had diverse regulation patterns to infect . GO function, involving cellular components, and KEGG pathways, involving glycerolipid metabolism, and plant-pathogen interaction were significantly enriched during infection by . The expression of ten genes in increased during the infection and infection stage, and these may regulate the response of to infection. This is the first comprehensive study on infection mechanism and response mechanism, which will lay a foundation for understanding the fungus-fungus interactions, gene functions, and variety breeding of pathogenic and edible fungi.
是羊肚菌的病原体之一,对羊肚菌的产量和品质有很大影响。在本研究中,我们组装了真菌SAAS_ppe1的基因组序列。我们分析了SAAS_ppe1在不同阶段(感染后3天和6天)感染羊肚菌的转录谱以及利用转录组分析羊肚菌的反应。SAAS_ppe1组装的基因组序列长度为39.78 Mb(11个支架;支架N50为6.50 Mb),其中检测到99.7%的预期基因。来自感染部位的清洁转录读数分别有7.48%和19.83%在感染的早期和晚期映射到羊肚菌基因组。在感染的不同阶段,羊肚菌中有3943个基因差异表达,其中24个基因随着感染和感染阶段表达增加,包括双氢酰胺生物合成、醛还原酶和NAD(P)H-水合表异构酶(P<0.05)。几个基因在感染的不同阶段有不同的表达趋势,表明羊肚菌对感染有多种调控模式。在SAAS_ppe1感染期间,涉及细胞成分的GO功能以及涉及甘油脂代谢和植物-病原体相互作用的KEGG途径显著富集。羊肚菌中十个基因的表达在感染和感染阶段增加,这些基因可能调节羊肚菌对SAAS_ppe1感染的反应。这是关于SAAS_ppe1感染机制和羊肚菌反应机制的首次全面研究,将为理解真菌-真菌相互作用、基因功能以及致病和食用真菌的品种育种奠定基础。