Department of Biotechnology and Food Science, NTNU - Norwegian University of Science and Technology, Trondheim, Norway.
K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and General Practice, NTNU - Norwegian University of Science and Technology, Trondheim, Norway.
PLoS Comput Biol. 2021 May 24;17(5):e1008528. doi: 10.1371/journal.pcbi.1008528. eCollection 2021 May.
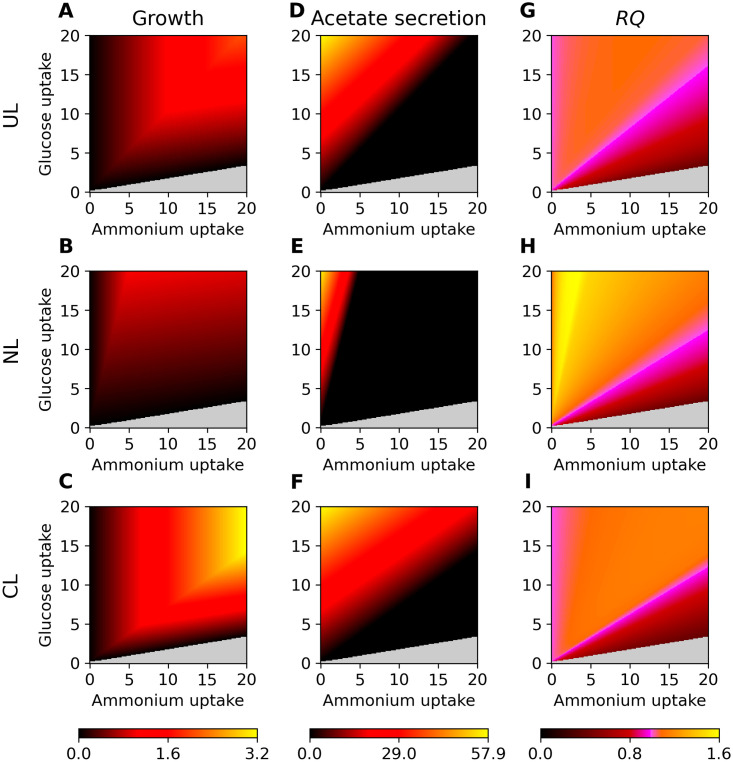
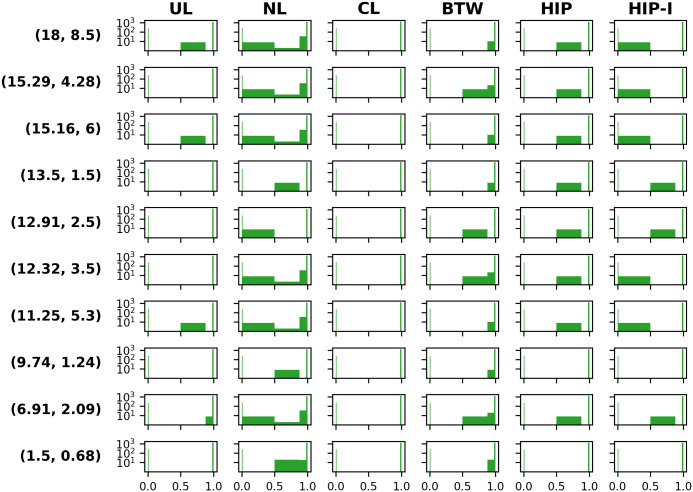
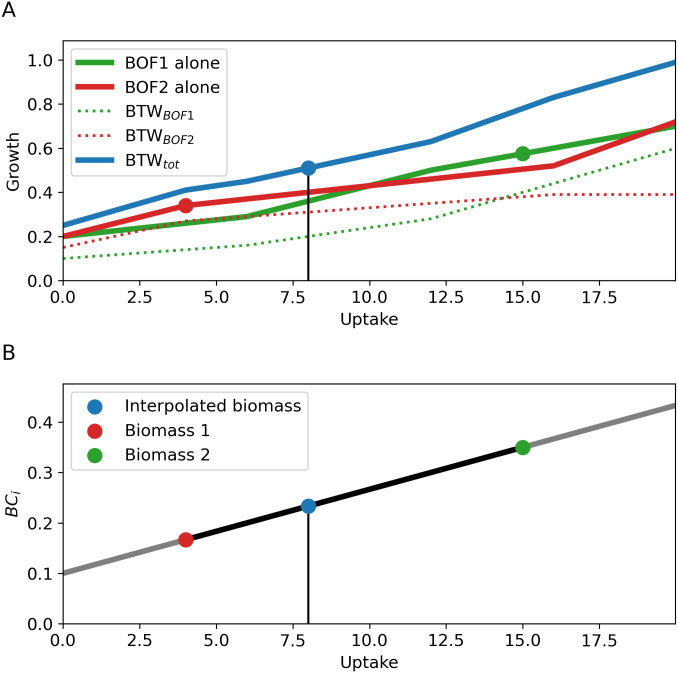
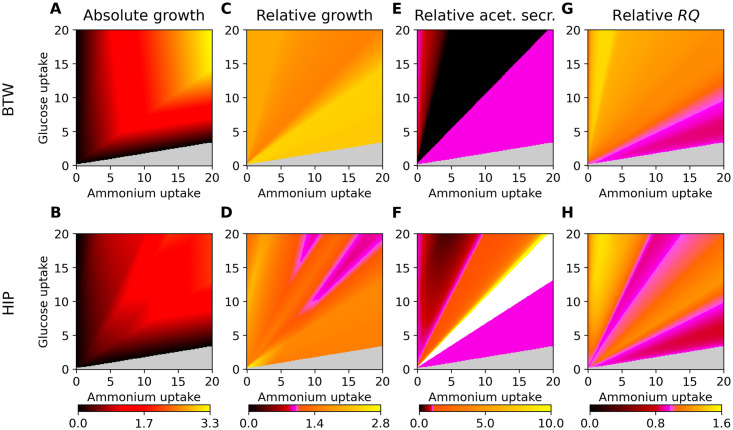
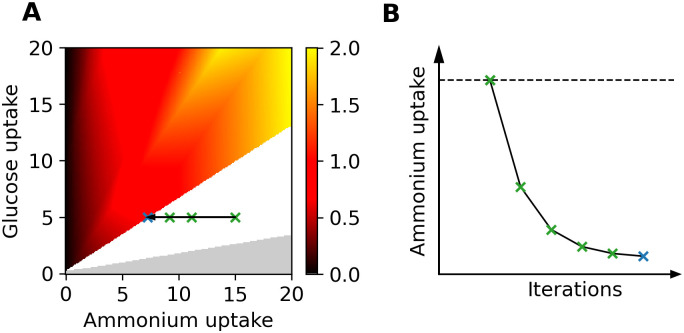
Genome-scale metabolic modeling is an important tool in the study of metabolism by enhancing the collation of knowledge, interpretation of data, and prediction of metabolic capabilities. A frequent assumption in the use of genome-scale models is that the in vivo organism is evolved for optimal growth, where growth is represented by flux through a biomass objective function (BOF). While the specific composition of the BOF is crucial, its formulation is often inherited from similar organisms due to the experimental challenges associated with its proper determination. A cell's macro-molecular composition is not fixed and it responds to changes in environmental conditions. As a consequence, initiatives for the high-fidelity determination of cellular biomass composition have been launched. Thus, there is a need for a mathematical and computational framework capable of using multiple measurements of cellular biomass composition in different environments. Here, we propose two different computational approaches for directly addressing this challenge: Biomass Trade-off Weighting (BTW) and Higher-dimensional-plane InterPolation (HIP). In lieu of experimental data on biomass composition-variation in response to changing nutrient environment, we assess the properties of BTW and HIP using three hypothetical, yet biologically plausible, BOFs for the Escherichia coli genome-scale metabolic model iML1515. We find that the BTW and HIP formulations have a significant impact on model performance and phenotypes. Furthermore, the BTW method generates larger growth rates in all environments when compared to HIP. Using acetate secretion and the respiratory quotient as proxies for phenotypic changes, we find marked differences between the methods as HIP generates BOFs more similar to a reference BOF than BTW. We conclude that the presented methods constitute a conceptual step in developing genome-scale metabolic modelling approaches capable of addressing the inherent dependence of cellular biomass composition on nutrient environments.
基因组规模代谢建模是代谢研究的重要工具,通过增强知识的整理、数据的解释和代谢能力的预测。在使用基因组规模模型时,一个常见的假设是,体内生物体会进化到最佳生长状态,生长表现为通过生物质目标函数(BOF)的通量。虽然 BOF 的具体组成至关重要,但由于其正确确定所涉及的实验挑战,其配方通常是从类似的生物体继承而来的。细胞的宏观分子组成不是固定的,它会对环境条件的变化做出反应。因此,已经发起了确定细胞生物质组成的高保真度的倡议。因此,需要一个能够在不同环境中使用细胞生物质组成的多个测量值的数学和计算框架。在这里,我们提出了两种不同的计算方法来直接解决这一挑战:生物质权衡加权(BTW)和高维平面内插(HIP)。在缺乏关于生物质组成随营养环境变化的实验数据的情况下,我们使用三种假设的、但具有生物学合理性的 BOF 来评估 BTW 和 HIP 的特性,这些 BOF 适用于大肠杆菌基因组规模代谢模型 iML1515。我们发现,BTW 和 HIP 配方对模型性能和表型有重大影响。此外,与 HIP 相比,BTW 方法在所有环境中都产生了更大的生长速率。使用乙酸分泌和呼吸商作为表型变化的代理,我们发现两种方法之间存在显著差异,因为 HIP 生成的 BOF 比 BTW 更类似于参考 BOF。我们得出的结论是,所提出的方法是朝着开发能够解决细胞生物质组成对营养环境固有依赖性的基因组规模代谢建模方法迈出的概念性一步。