Karnan Sivasundaram, Hanamura Ichiro, Ota Akinobu, Takasugi Souichi, Nakamura Ayano, Takahashi Miyuki, Uchino Kaori, Murakami Satsuki, Wahiduzzaman Md, Quang Vu Lam, Rahman Md Lutfur, Hasan Muhammad Nazmul, Hyodo Toshinori, Konishi Hiroyuki, Tsuzuki Shinobu, Yoshikawa Kazuhiro, Suzuki Susumu, Ueda Ryuzo, Ejiri Masayuki, Hosokawa Yoshitaka, Takami Akiyoshi
Department of Biochemistry, Aichi Medical University, Nagakute, Aichi, Japan.
Division of Hematology, Department of Internal Medicine, Aichi Medical University, Nagakute, Aichi, Japan.
Cell Death Discov. 2021 May 25;7(1):121. doi: 10.1038/s41420-021-00446-8.
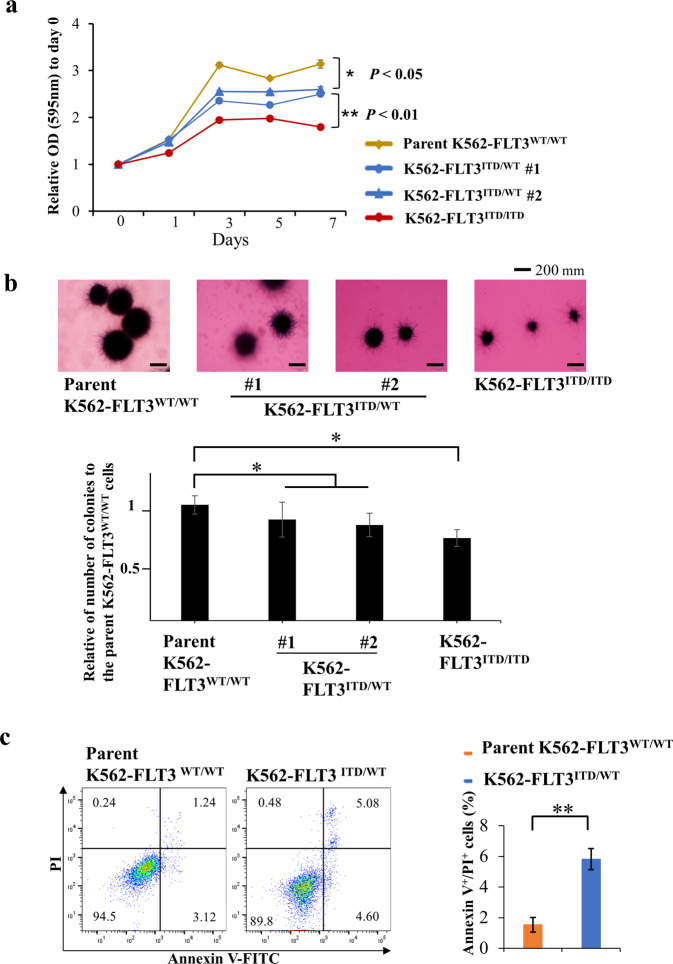
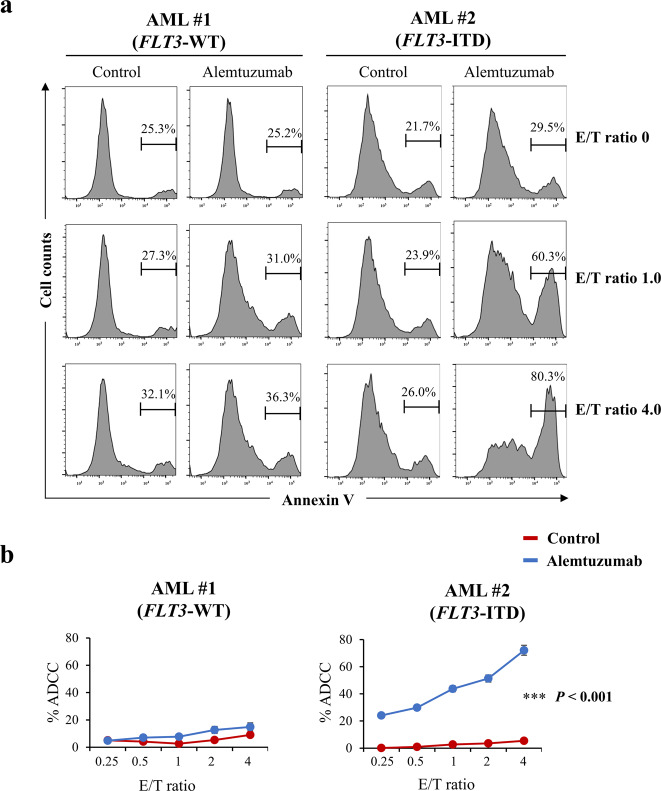
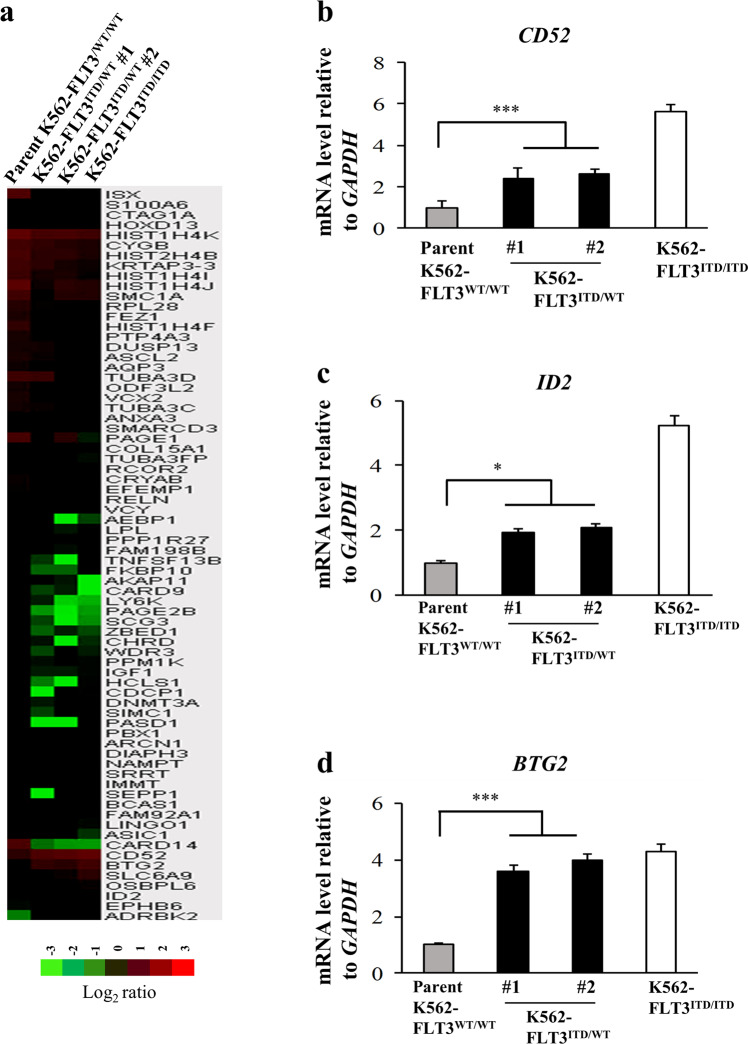
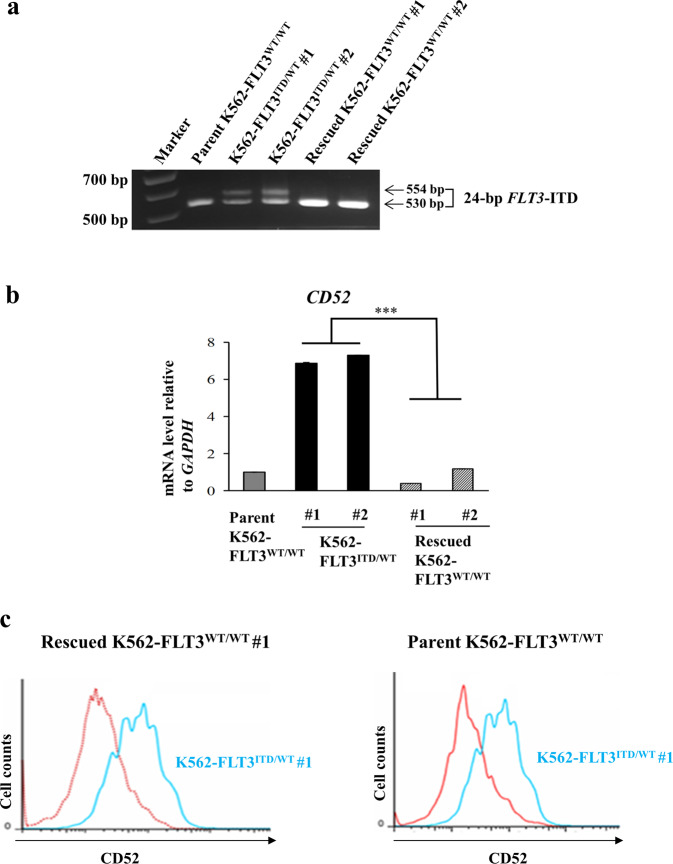
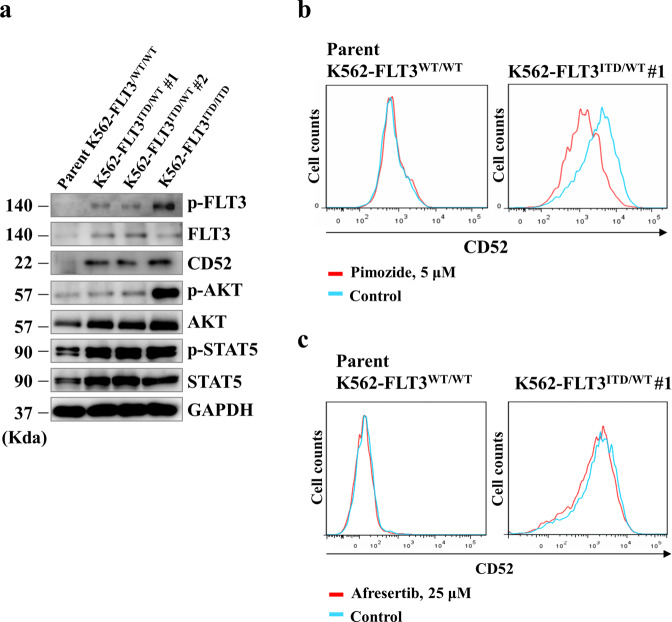
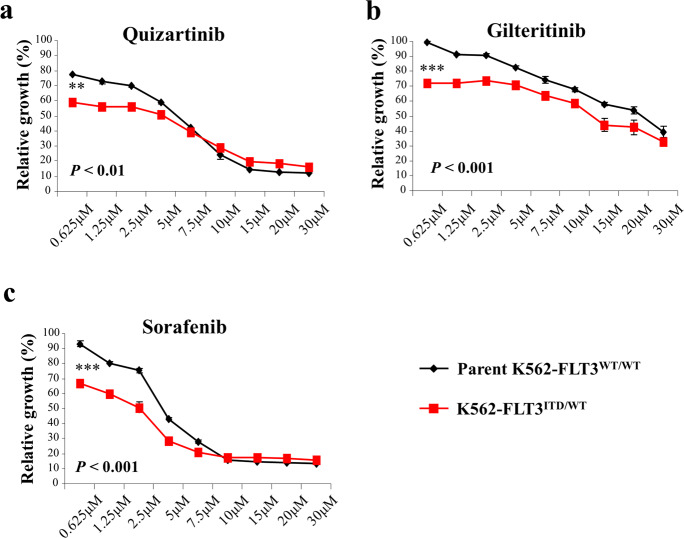
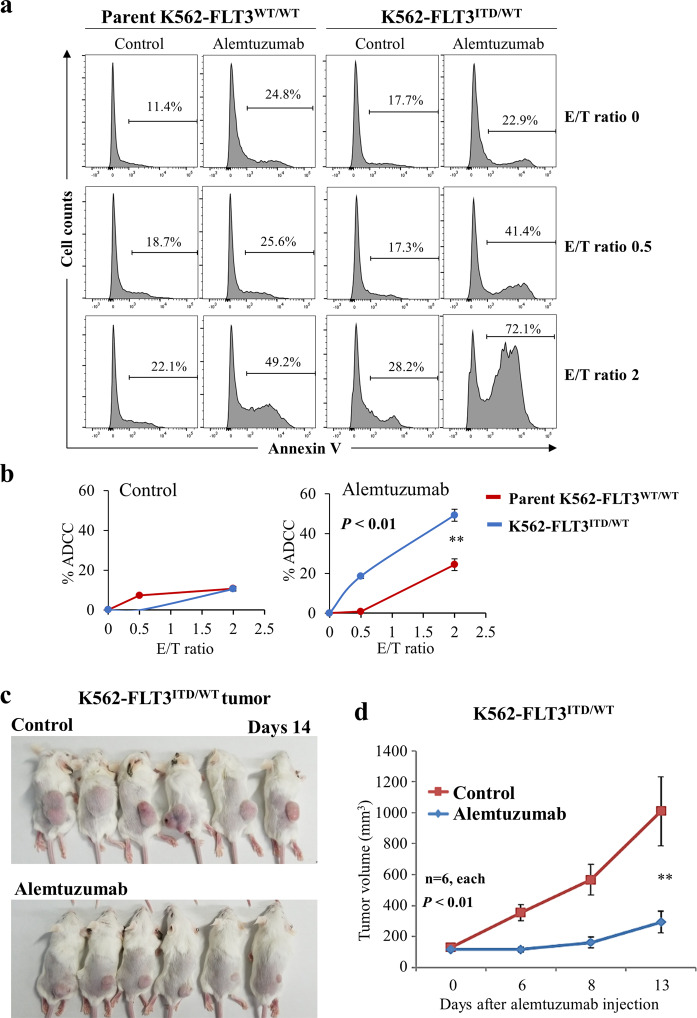
Internal tandem duplication (ITD) of FMS-like tyrosine kinase 3 (FLT3) confers poor prognosis and is found in approximately 25% of cases of acute myeloid leukemia (AML). Although FLT3 inhibitors have shown clinical benefit in patients with AML harboring FLT3-ITD, the therapeutic effect is limited. Here, to explore alternative therapeutics, we established a cellular model of monoallelic FLT3 cells using the CRISPR-Cas9 system in a human myeloid leukemia cell line, K562. cDNA microarray analysis revealed elevated CD52 expression in K562-FLT3 cells compared to K562-FLT3 cells, an observation that was further confirmed by quantitative real-time-PCR and flow cytometric analyses. The elevated expression of CD52 in K562-FLT3 cells was decreased in wild-type FLT3 (FLT3-WT) knock-in K562-FLT3 cells. In K562-FLT3 cells, a STAT5 inhibitor, pimozide, downregulated CD52 protein expression while an AKT inhibitor, afuresertib, did not affect CD52 expression. Notably, an anti-CD52 antibody, alemtuzumab, induced significant antibody-dependent cell-mediated cytotoxicity (ADCC) in K562-FLT3 cells compared to K562-FLT3 cells. Furthermore, alemtuzumab significantly suppressed the xenograft tumor growth of K562-FLT3 cells in severe combined immunodeficiency (SCID) mice. Taken together, our data suggested that genetically modified FLT3-ITD knock-in human myeloid leukemia K562 cells upregulated CD52 expression via activation of STAT5, and alemtuzumab showed an antitumor effect via induction of ADCC in K562-FLT3 cells. Our findings may allow establishment of a new therapeutic option, alemtuzumab, to treat leukemia with the FLT3-ITD mutation.
FMS样酪氨酸激酶3(FLT3)的内部串联重复(ITD)预示着预后不良,约25%的急性髓系白血病(AML)病例中可检测到该突变。尽管FLT3抑制剂已在携带FLT3-ITD的AML患者中显示出临床疗效,但治疗效果有限。在此,为探索替代疗法,我们利用CRISPR-Cas9系统在人髓系白血病细胞系K562中建立了单等位基因FLT3细胞的细胞模型。cDNA微阵列分析显示,与K562-FLT3细胞相比,K562-FLT3细胞中CD52表达升高,这一观察结果通过定量实时PCR和流式细胞术分析得到进一步证实。野生型FLT3(FLT3-WT)敲入的K562-FLT3细胞中,K562-FLT3细胞中升高的CD52表达降低。在K562-FLT3细胞中,STAT5抑制剂匹莫齐特下调CD52蛋白表达,而AKT抑制剂阿福司他不影响CD52表达。值得注意的是,与K562-FLT3细胞相比,抗CD52抗体阿仑单抗在K562-FLT3细胞中诱导了显著的抗体依赖性细胞介导的细胞毒性(ADCC)。此外,阿仑单抗显著抑制了严重联合免疫缺陷(SCID)小鼠中K562-FLT3细胞的异种移植肿瘤生长。综上所述,我们的数据表明,基因改造的FLT3-ITD敲入人髓系白血病K562细胞通过激活STAT5上调CD52表达,阿仑单抗通过在K562-FLT3细胞中诱导ADCC显示出抗肿瘤作用。我们的研究结果可能有助于建立一种新的治疗选择——阿仑单抗,用于治疗携带FLT3-ITD突变的白血病。