San James E, Ngcapu Sinaye, Kanzi Aquillah M, Tegally Houriiyah, Fonseca Vagner, Giandhari Jennifer, Wilkinson Eduan, Nelson Chase W, Smidt Werner, Kiran Anmol M, Chimukangara Benjamin, Pillay Sureshnee, Singh Lavanya, Fish Maryam, Gazy Inbal, Martin Darren P, Khanyile Khulekani, Lessells Richard, de Oliveira Tulio
KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine & Medical Sciences, University of KwaZulu- Natal, Durban, South Africa.
Centre for the AIDS Programme of Research in South Africa (CAPRISA), Durban, South Africa.
Virus Evol. 2021 Apr 21;7(1):veab041. doi: 10.1093/ve/veab041. eCollection 2021 Jan.
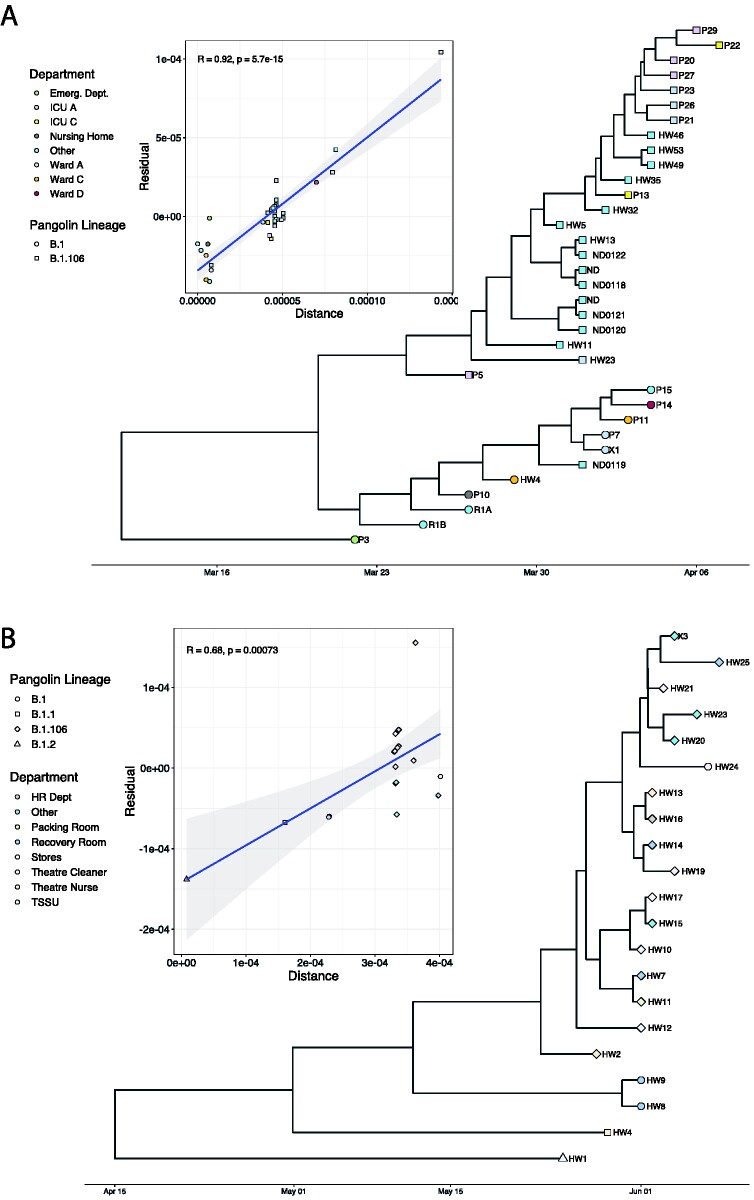
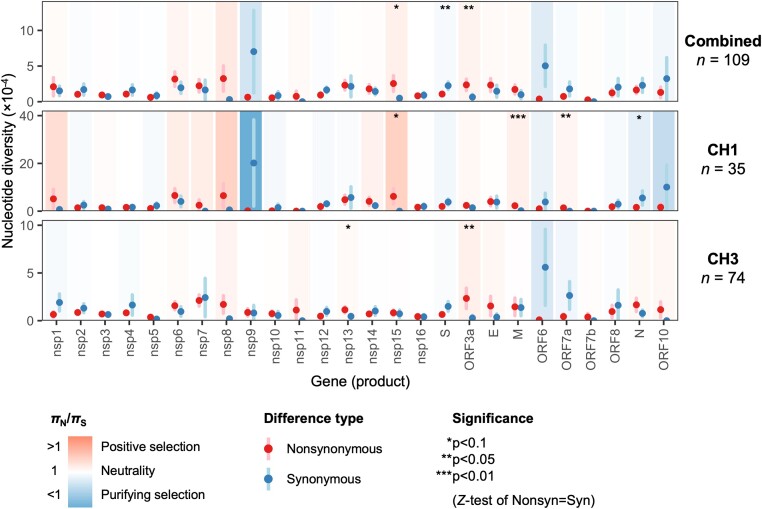
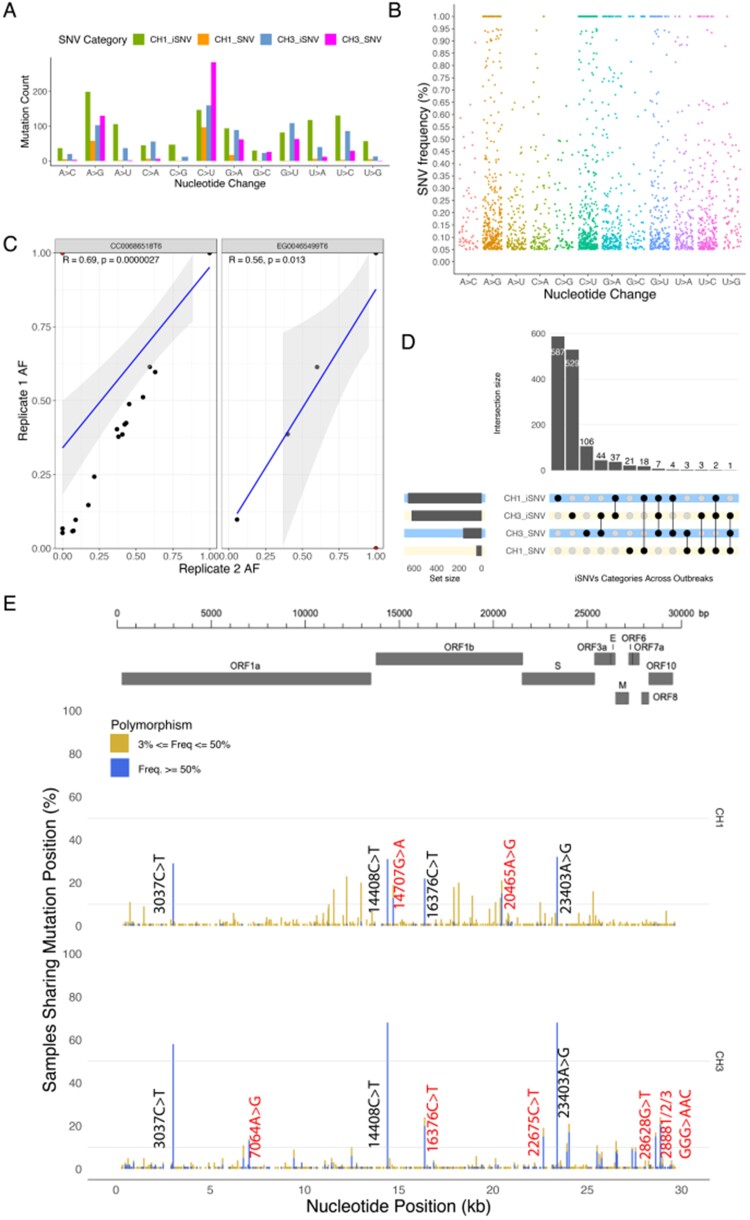
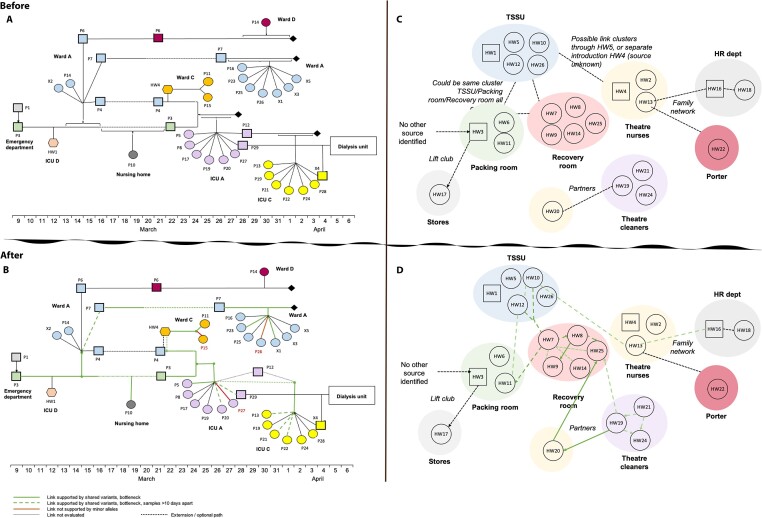
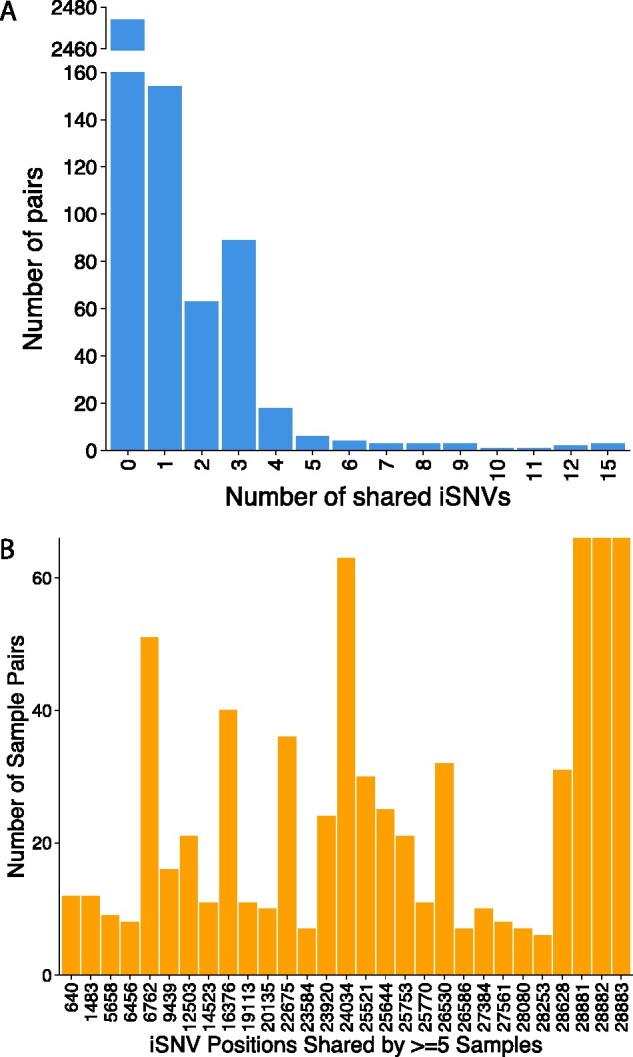
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes acute, highly transmissible respiratory infection in humans and a wide range of animal species. Its rapid global spread has resulted in a major public health emergency, necessitating commensurately rapid research to improve control strategies. In particular, the ability to effectively retrace transmission chains in outbreaks remains a major challenge, partly due to our limited understanding of the virus' underlying evolutionary dynamics within and between hosts. We used high-throughput sequencing whole-genome data coupled with bottleneck analysis to retrace the pathways of viral transmission in two nosocomial outbreaks that were previously characterised by epidemiological and phylogenetic methods. Additionally, we assessed the mutational landscape, selection pressures, and diversity at the within-host level for both outbreaks. Our findings show evidence of within-host selection and transmission of variants between samples. Both bottleneck and diversity analyses highlight within-host and consensus-level variants shared by putative source-recipient pairs in both outbreaks, suggesting that certain within-host variants in these outbreaks may have been transmitted upon infection rather than arising independently within multiple hosts. Overall, our findings demonstrate the utility of combining within-host diversity and bottleneck estimations for elucidating transmission events in SARS-CoV-2 outbreaks, provide insight into the maintenance of viral genetic diversity, provide a list of candidate targets of positive selection for further investigation, and demonstrate that within-host variants can be transferred between patients. Together these results will help in developing strategies to understand the nature of transmission events and curtail the spread of SARS-CoV-2.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)可导致人类及多种动物发生急性、高传染性呼吸道感染。其在全球的迅速传播已引发重大突发公共卫生事件,因此需要开展相应快速的研究以改进防控策略。特别是,在疫情中有效追溯传播链的能力仍然是一项重大挑战,部分原因在于我们对该病毒在宿主内部及宿主之间的潜在进化动态了解有限。我们利用高通量测序全基因组数据并结合瓶颈分析,来追溯此前通过流行病学和系统发育方法表征的两起医院内疫情中的病毒传播途径。此外,我们评估了这两起疫情在宿主内水平的突变格局、选择压力和多样性。我们的研究结果显示存在宿主内选择以及样本间变异传播的证据。瓶颈分析和多样性分析均突出了两起疫情中假定的源-受体对所共有的宿主内及共识水平变异,这表明这些疫情中的某些宿主内变异可能在感染时就已传播,而非在多个宿主内独立产生。总体而言,我们的研究结果证明了结合宿主内多样性和瓶颈估计来阐明SARS-CoV-2疫情中传播事件的实用性,深入了解了病毒遗传多样性的维持情况,提供了一系列有待进一步研究的正选择候选靶点,并证明了宿主内变异可在患者之间传播。这些结果共同将有助于制定策略以了解传播事件的本质并遏制SARS-CoV-2的传播。