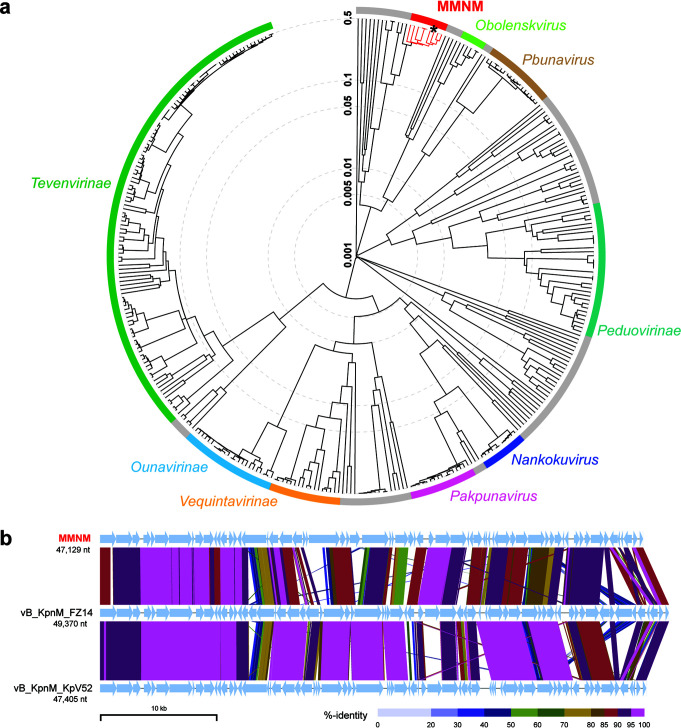
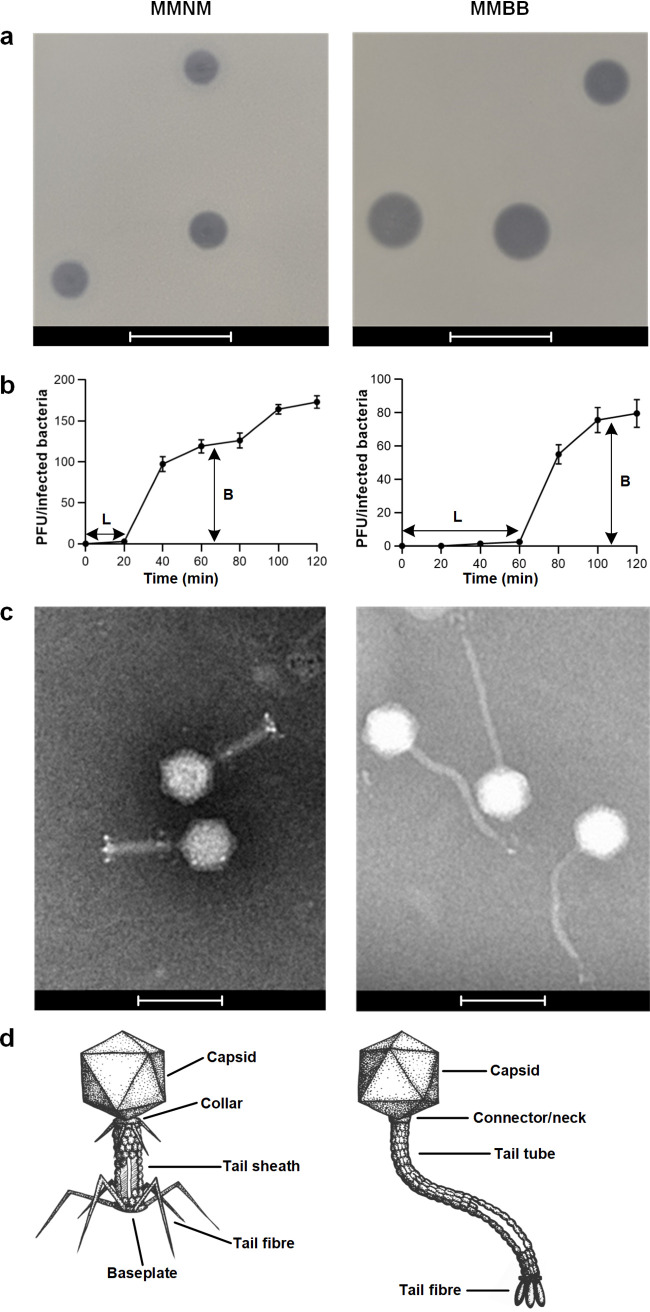
Thung Tze Y, White Murray E, Dai Wei, Wilksch Jonathan J, Bamert Rebecca S, Rocker Andrea, Stubenrauch Christopher J, Williams Daniel, Huang Cheng, Schittelhelm Ralf, Barr Jeremy J, Jameson Eleanor, McGowan Sheena, Zhang Yanju, Wang Jiawei, Dunstan Rhys A, Lithgow Trevor
Infection & Immunity Program, Biomedicine Discovery Institute, Monash University, Clayton, Australia.
Department of Microbiology, Monash University, Clayton, Australia.
mSystems. 2021 Jun 29;6(3):e0024221. doi: 10.1128/mSystems.00242-21. Epub 2021 May 27.
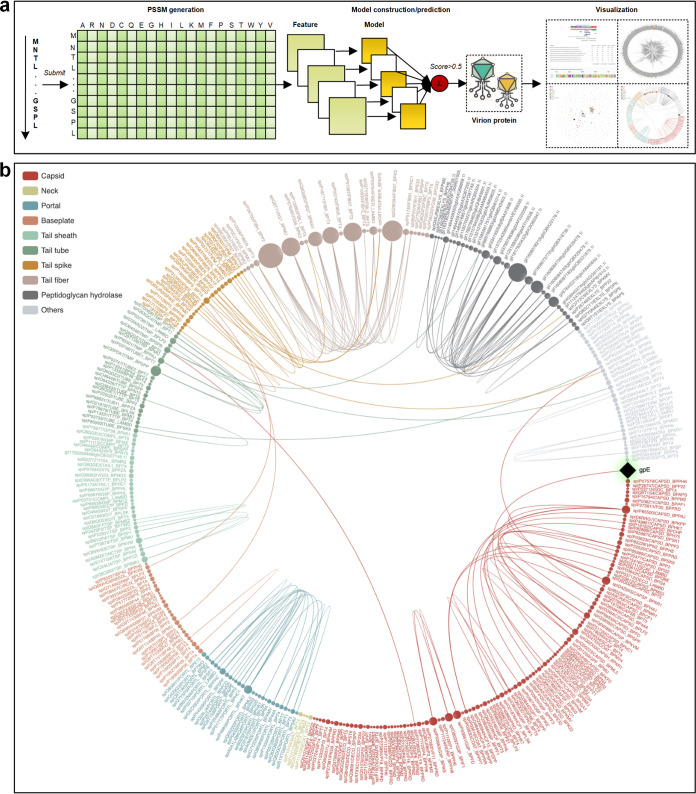
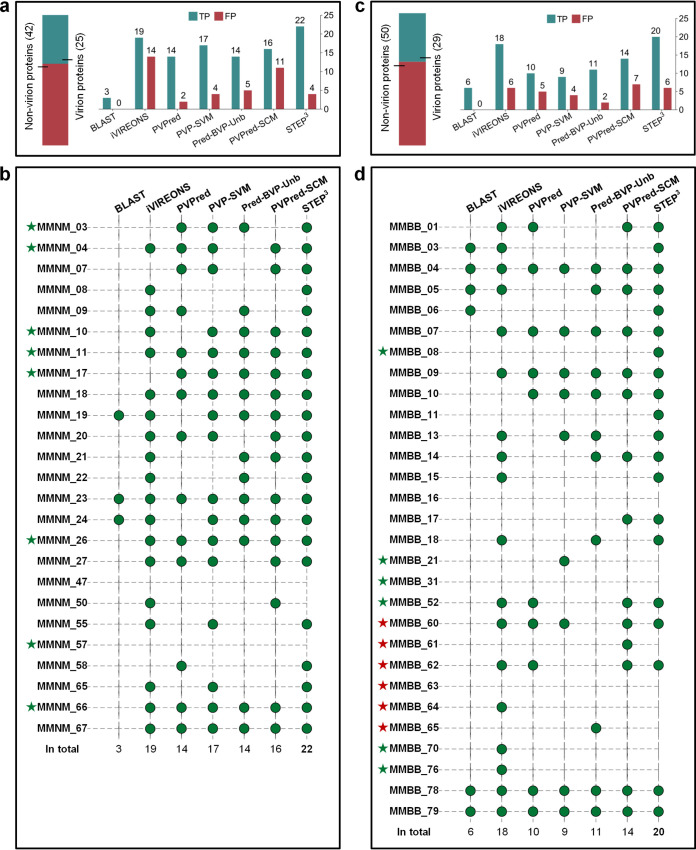
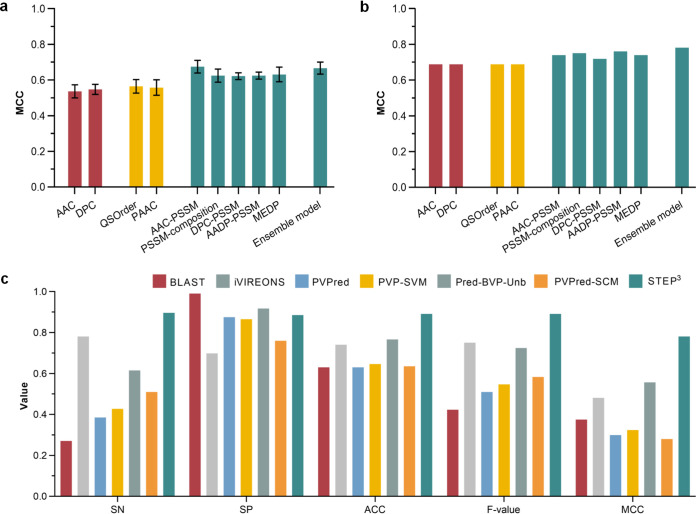
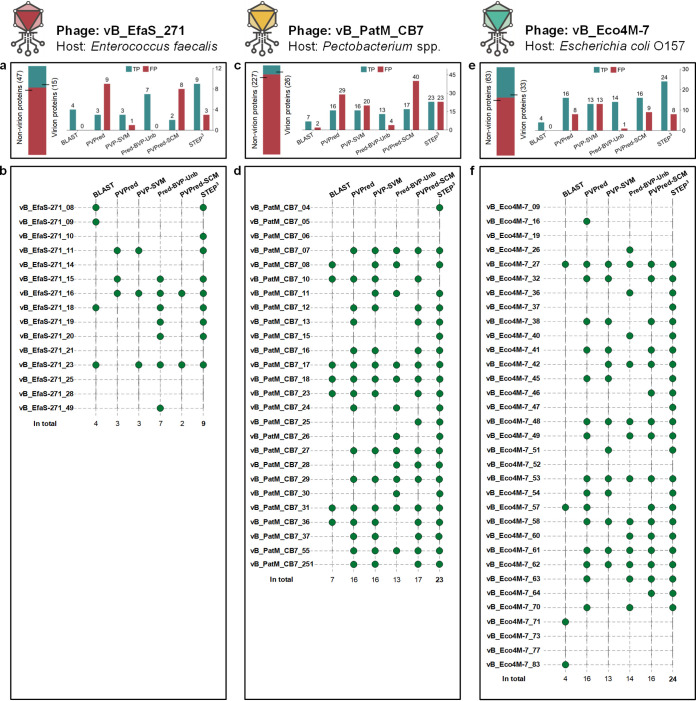
Antimicrobial resistance (AMR) continues to evolve as a major threat to human health, and new strategies are required for the treatment of AMR infections. Bacteriophages (phages) that kill bacterial pathogens are being identified for use in phage therapies, with the intention to apply these bactericidal viruses directly into the infection sites in bespoke phage cocktails. Despite the great unsampled phage diversity for this purpose, an issue hampering the roll out of phage therapy is the poor quality annotation of many of the phage genomes, particularly for those from infrequently sampled environmental sources. We developed a computational tool called STEP to use the "evolutionary features" that can be recognized in genome sequences of diverse phages. These features, when integrated into an ensemble framework, achieved a stable and robust prediction performance when benchmarked against other prediction tools using phages from diverse sources. Validation of the prediction accuracy of STEP was conducted with high-resolution mass spectrometry analysis of two novel phages, isolated from a watercourse in the Southern Hemisphere. STEP provides a robust computational approach to distinguish specific and universal features in phages to improve the quality of phage cocktails and is available for use at http://step3.erc.monash.edu/. In response to the global problem of antimicrobial resistance, there are moves to use bacteriophages (phages) as therapeutic agents. Selecting which phages will be effective therapeutics relies on interpreting features contributing to shelf-life and applicability to diagnosed infections. However, the protein components of the phage virions that dictate these properties vary so much in sequence that best estimates suggest failure to recognize up to 90% of them. We have utilized this diversity in evolutionary features as an advantage, to apply machine learning for prediction accuracy for diverse components in phage virions. We benchmark this new tool showing the accurate recognition and evaluation of phage component parts using genome sequence data of phages from undersampled environments, where the richest diversity of phage still lies.
抗菌耐药性(AMR)持续演变,成为对人类健康的重大威胁,因此需要新的策略来治疗AMR感染。能杀死细菌病原体的噬菌体正被确定用于噬菌体疗法,目的是将这些杀菌病毒直接以定制的噬菌体鸡尾酒形式应用于感染部位。尽管为此目的存在大量未采样的噬菌体多样性,但阻碍噬菌体疗法推广的一个问题是许多噬菌体基因组的注释质量较差,尤其是那些来自采样较少的环境来源的噬菌体。我们开发了一种名为STEP的计算工具,以利用在不同噬菌体的基因组序列中可识别的“进化特征”。当与使用来自不同来源的噬菌体的其他预测工具进行基准测试时,这些特征整合到一个集成框架中,实现了稳定且强大的预测性能。通过对从南半球一条水道分离出的两种新型噬菌体进行高分辨率质谱分析,对STEP的预测准确性进行了验证。STEP提供了一种强大的计算方法,以区分噬菌体中的特定和通用特征,从而提高噬菌体鸡尾酒的质量,可在http://step3.erc.monash.edu/上使用。针对全球抗菌耐药性问题,人们正在采取行动将噬菌体用作治疗剂。选择哪些噬菌体将成为有效的治疗剂依赖于解读有助于保质期和对已诊断感染适用性的特征。然而,决定这些特性 的噬菌体病毒粒子的蛋白质成分在序列上差异极大,最佳估计表明多达90%的成分无法识别。我们利用这种进化特征的多样性作为优势应用机器学习来预测噬菌体病毒粒子中不同成分的准确性。我们使用来自采样不足的环境(其中噬菌体的多样性最为丰富)的噬菌体基因组序列数据对这个新工具进行基准测试,展示了对噬菌体组成部分的准确识别和评估。