Interuniversity Institute of Bioinformatics in Brussels, Université Libre de Bruxelles, Brussels, Belgium.
BMC Bioinformatics. 2021 May 27;22(1):280. doi: 10.1186/s12859-021-04210-8.
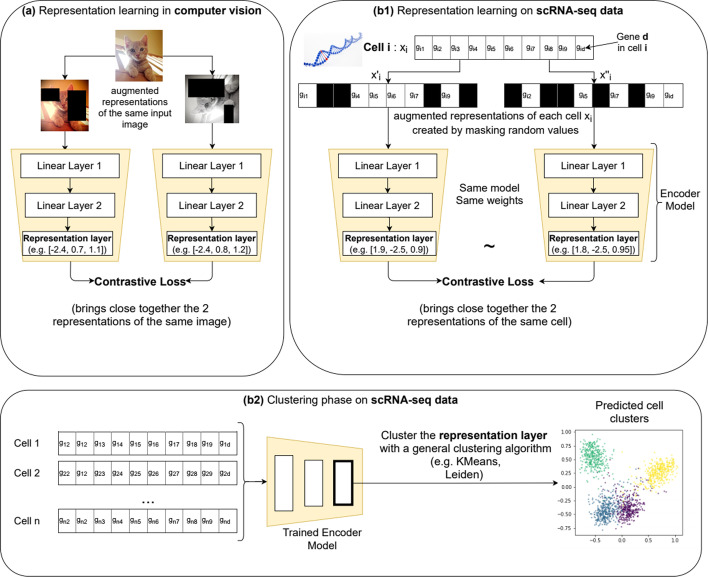
Single-cell RNA sequencing (scRNA-seq) has emerged has a main strategy to study transcriptional activity at the cellular level. Clustering analysis is routinely performed on scRNA-seq data to explore, recognize or discover underlying cell identities. The high dimensionality of scRNA-seq data and its significant sparsity accentuated by frequent dropout events, introducing false zero count observations, make the clustering analysis computationally challenging. Even though multiple scRNA-seq clustering techniques have been proposed, there is no consensus on the best performing approach. On a parallel research track, self-supervised contrastive learning recently achieved state-of-the-art results on images clustering and, subsequently, image classification.
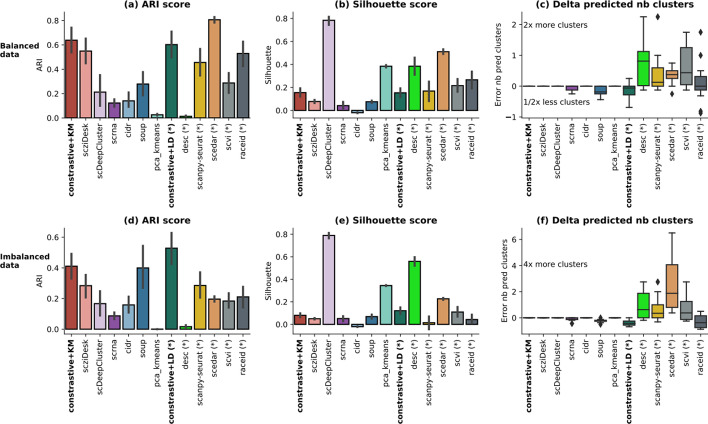
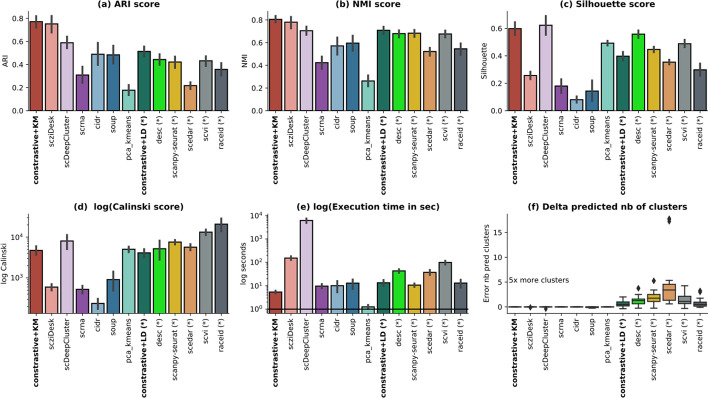
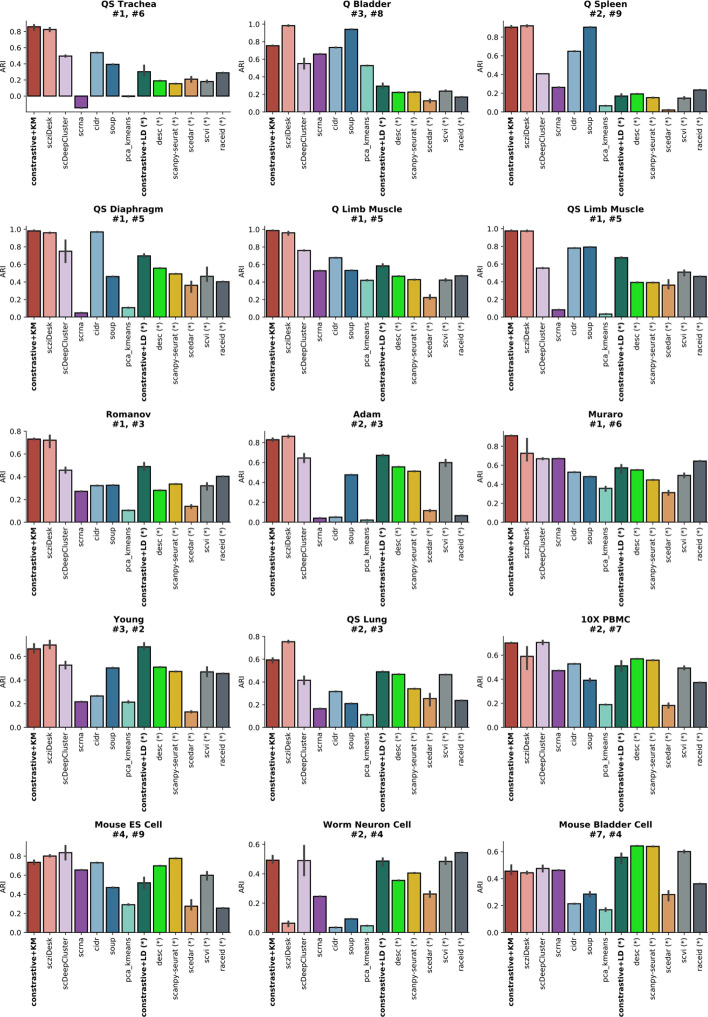
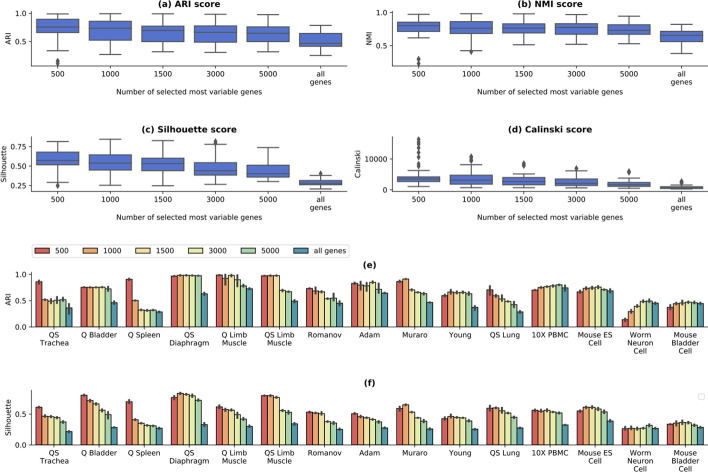
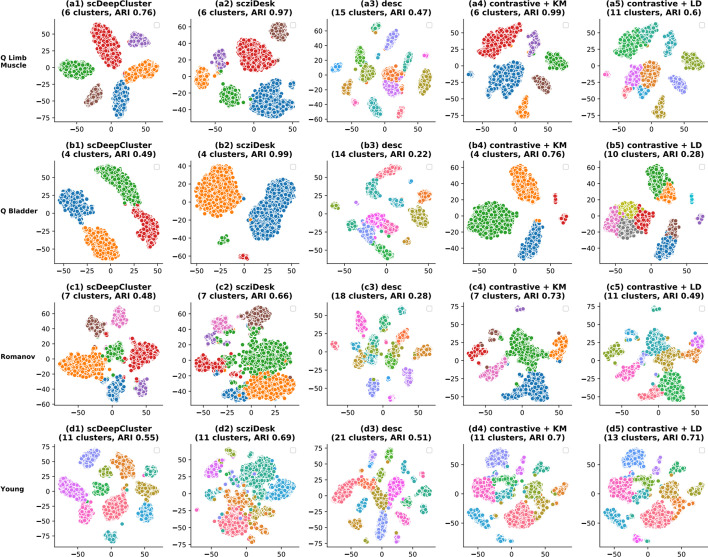
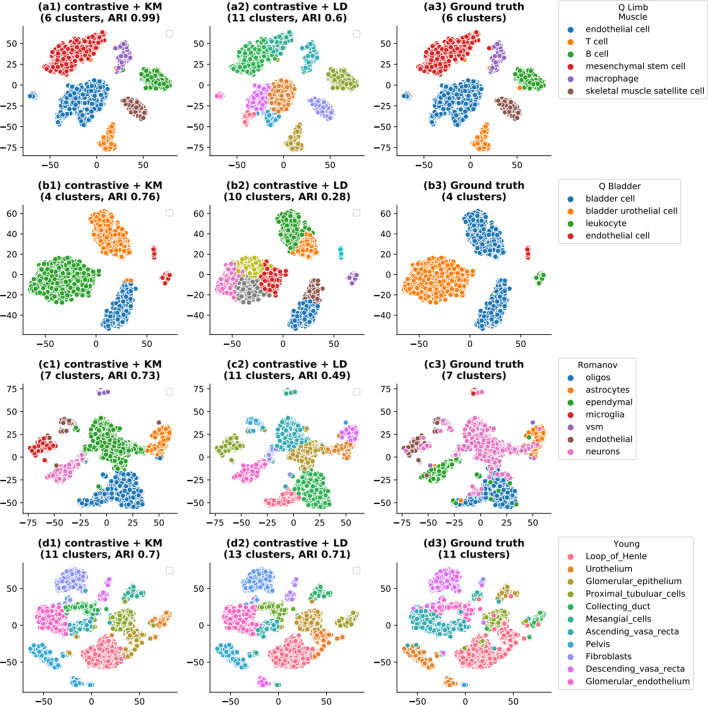
We propose contrastive-sc, a new unsupervised learning method for scRNA-seq data that perform cell clustering. The method consists of two consecutive phases: first, an artificial neural network learns an embedding for each cell through a representation training phase. The embedding is then clustered in the second phase with a general clustering algorithm (i.e. KMeans or Leiden community detection). The proposed representation training phase is a new adaptation of the self-supervised contrastive learning framework, initially proposed for image processing, to scRNA-seq data. contrastive-sc has been compared with ten state-of-the-art techniques. A broad experimental study has been conducted on both simulated and real-world datasets, assessing multiple external and internal clustering performance metrics (i.e. ARI, NMI, Silhouette, Calinski scores). Our experimental analysis shows that constastive-sc compares favorably with state-of-the-art methods on both simulated and real-world datasets.
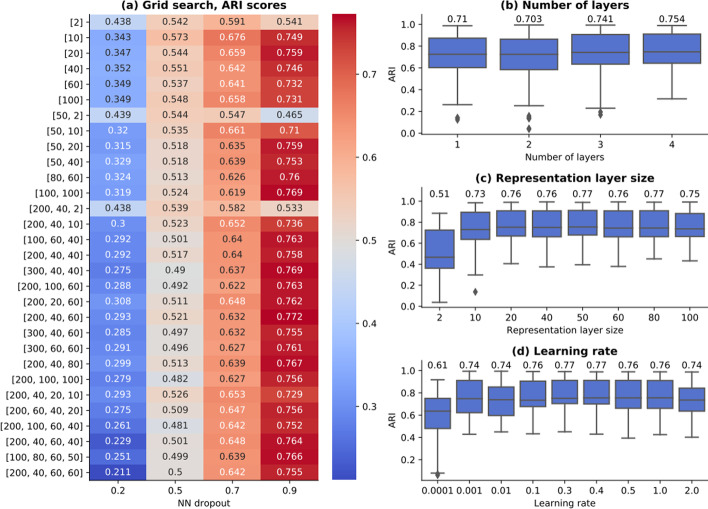
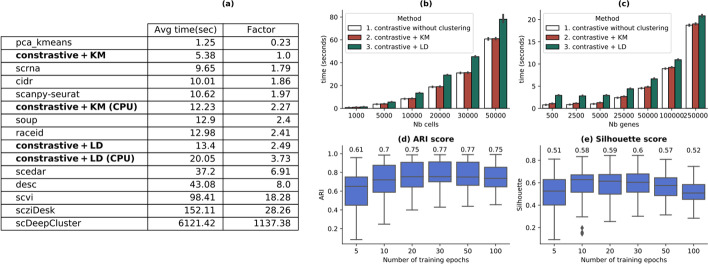
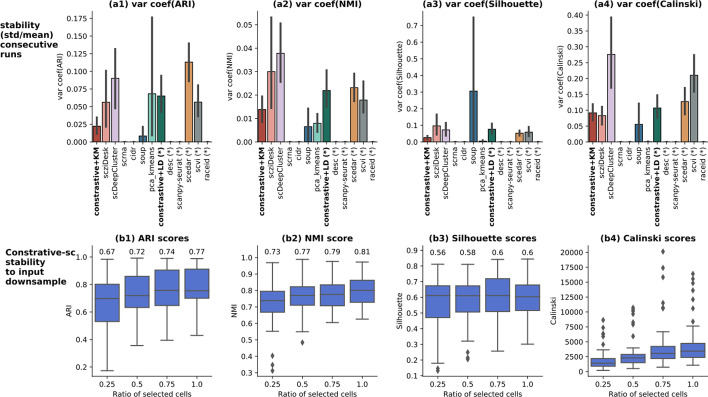
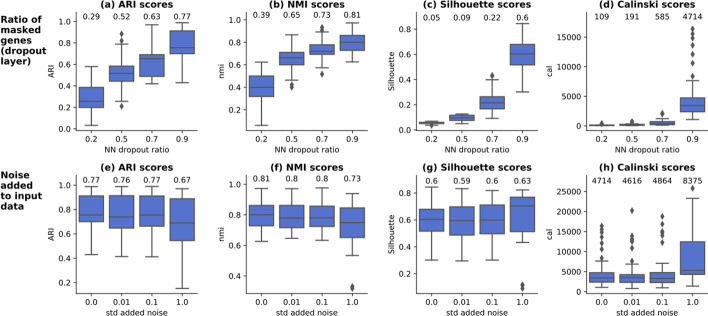
On average, our method identifies well-defined clusters in close agreement with ground truth annotations. Our method is computationally efficient, being fast to train and having a limited memory footprint. contrastive-sc maintains good performance when only a fraction of input cells is provided and is robust to changes in hyperparameters or network architecture. The decoupling between the creation of the embedding and the clustering phase allows the flexibility to choose a suitable clustering algorithm (i.e. KMeans when the number of expected clusters is known, Leiden otherwise) or to integrate the embedding with other existing techniques.
单细胞 RNA 测序 (scRNA-seq) 已成为研究细胞水平转录活性的主要策略。聚类分析通常在 scRNA-seq 数据上进行,以探索、识别或发现潜在的细胞身份。scRNA-seq 数据的高维度及其由于频繁的缺失事件而显著稀疏,引入了虚假的零计数观察值,使得聚类分析具有计算挑战性。尽管已经提出了多种 scRNA-seq 聚类技术,但对于最佳表现方法尚无共识。在平行的研究轨道上,自监督对比学习最近在图像聚类方面取得了最先进的结果,随后在图像分类方面也取得了最先进的结果。
我们提出了 contrastive-sc,这是一种用于 scRNA-seq 数据的新无监督学习方法,可进行细胞聚类。该方法由两个连续的阶段组成:首先,通过表示训练阶段,人工神经网络为每个细胞学习一个嵌入。然后,在第二阶段,使用通用聚类算法(即 KMeans 或 Leiden 社区检测)对嵌入进行聚类。所提出的表示训练阶段是一种自我监督对比学习框架的新适应,最初是为图像处理提出的,现在也适用于 scRNA-seq 数据。contrastive-sc 已与十种最先进的技术进行了比较。在模拟和真实数据集上进行了广泛的实验研究,评估了多种外部和内部聚类性能指标(即 ARI、NMI、Silhouette、Calinski 分数)。我们的实验分析表明,在模拟和真实数据集上,contrastive-sc 与最先进的方法相比表现出色。
平均而言,我们的方法可以识别定义明确的聚类,与地面真实注释非常吻合。我们的方法计算效率高,训练速度快,内存占用有限。当仅提供输入细胞的一小部分时,contrastive-sc 保持良好的性能,并且对超参数或网络架构的变化具有鲁棒性。嵌入的创建和解耦与聚类阶段的分离允许灵活选择合适的聚类算法(即当已知预期聚类数时选择 KMeans,否则选择 Leiden),或者将嵌入与其他现有技术集成。