Song Baoxing, Buckler Edward S, Wang Hai, Wu Yaoyao, Rees Evan, Kellogg Elizabeth A, Gates Daniel J, Khaipho-Burch Merritt, Bradbury Peter J, Ross-Ibarra Jeffrey, Hufford Matthew B, Romay M Cinta
Institute for Genomic Diversity, Cornell University, Ithaca, New York 14853, USA.
Section of Plant Breeding and Genetics, Cornell University, Ithaca, New York 14853, USA.
Genome Res. 2021 Jul;31(7):1245-1257. doi: 10.1101/gr.266528.120. Epub 2021 May 27.
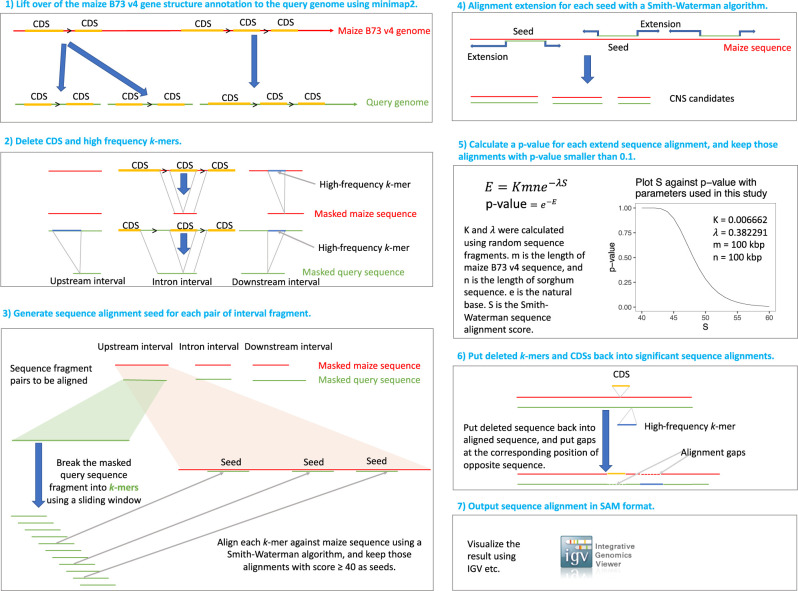
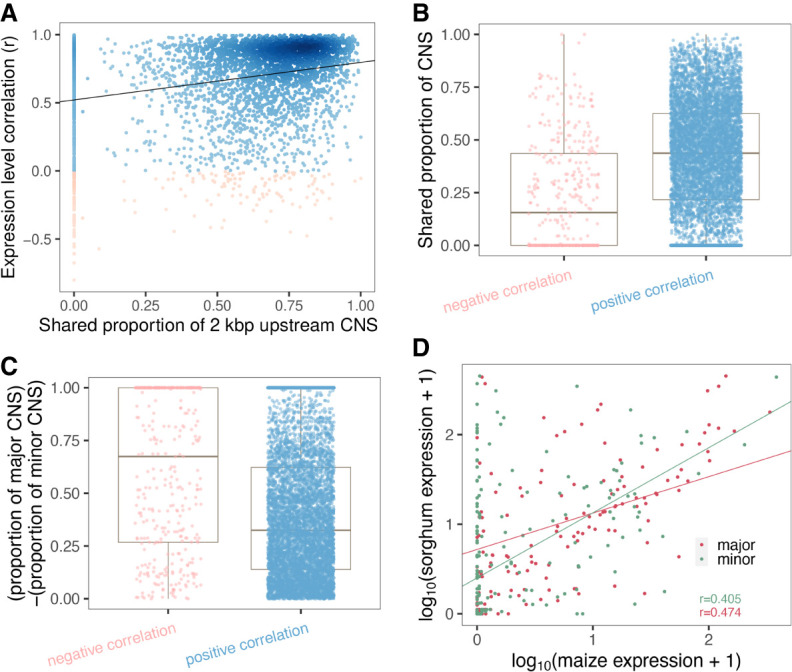
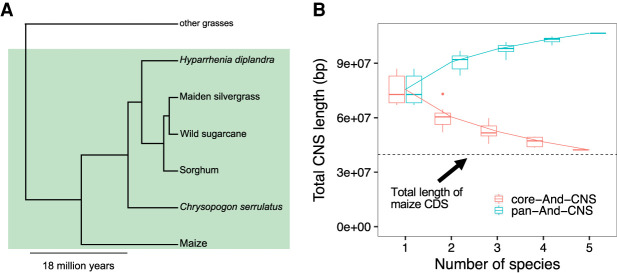
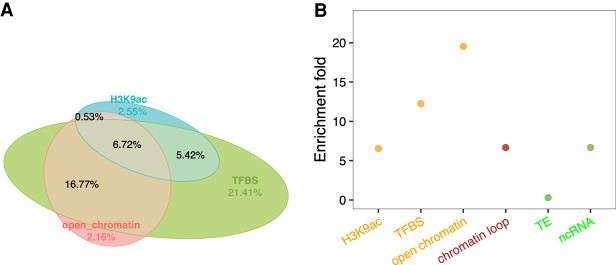
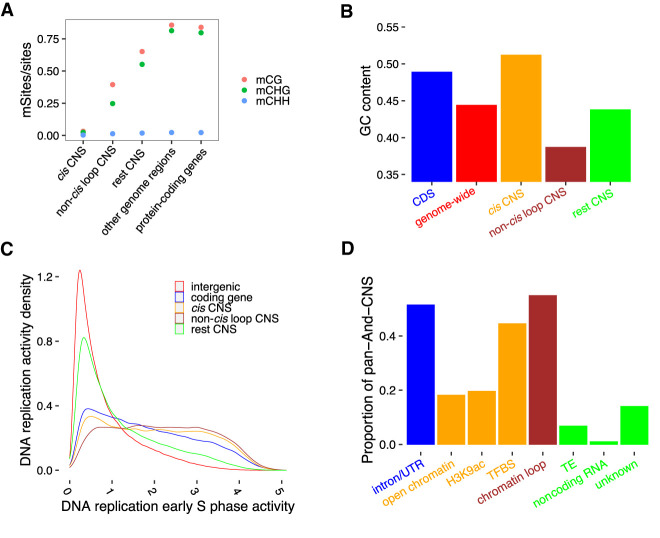
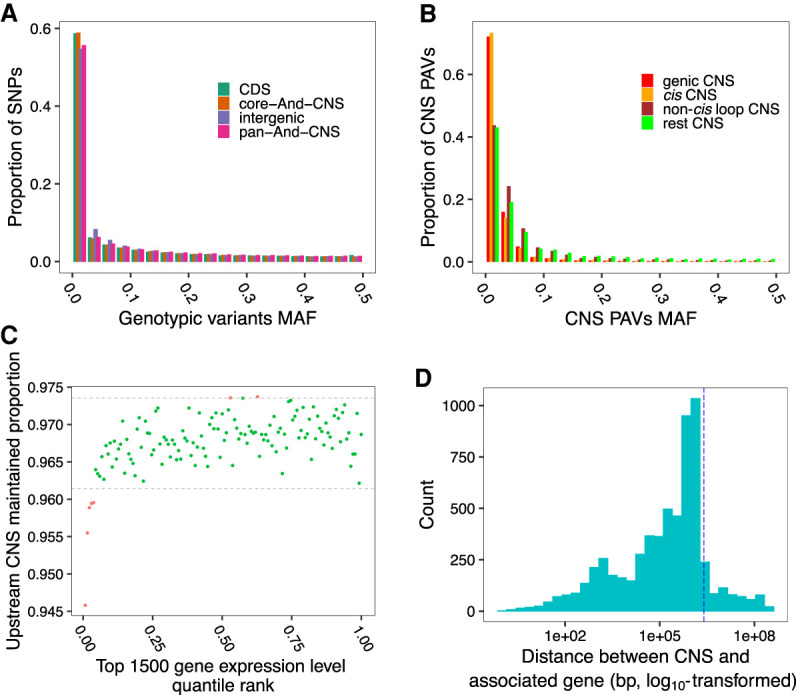
Thousands of species will be sequenced in the next few years; however, understanding how their genomes work, without an unlimited budget, requires both molecular and novel evolutionary approaches. We developed a sensitive sequence alignment pipeline to identify conserved noncoding sequences (CNSs) in the Andropogoneae tribe (multiple crop species descended from a common ancestor ∼18 million years ago). The Andropogoneae share similar physiology while being tremendously genomically diverse, harboring a broad range of ploidy levels, structural variation, and transposons. These contribute to the potential of Andropogoneae as a powerful system for studying CNSs and are factors we leverage to understand the function of maize CNSs. We found that 86% of CNSs were comprised of annotated features, including introns, UTRs, putative -regulatory elements, chromatin loop anchors, noncoding RNA (ncRNA) genes, and several transposable element superfamilies. CNSs were enriched in active regions of DNA replication in the early S phase of the mitotic cell cycle and showed different DNA methylation ratios compared to the genome-wide background. More than half of putative -regulatory sequences (identified via other methods) overlapped with CNSs detected in this study. Variants in CNSs were associated with gene expression levels, and CNS absence contributed to loss of gene expression. Furthermore, the evolution of CNSs was associated with the functional diversification of duplicated genes in the context of maize subgenomes. Our results provide a quantitative understanding of the molecular processes governing the evolution of CNSs in maize.
在未来几年内,将会有成千上万种物种进行测序;然而,在预算有限的情况下,要理解它们的基因组如何运作,就需要分子生物学方法和新颖的进化方法。我们开发了一种灵敏的序列比对流程,以识别黍族(约1800万年前起源于一个共同祖先的多个作物物种)中的保守非编码序列(CNS)。黍族具有相似的生理特征,但其基因组却极为多样,包含广泛的倍性水平、结构变异和转座子。这些因素使得黍族成为研究CNS的强大系统,我们利用这些因素来理解玉米CNS的功能。我们发现,86%的CNS由注释特征组成,包括内含子、非翻译区、假定的调控元件、染色质环锚、非编码RNA(ncRNA)基因以及几个转座子超家族。CNS在有丝分裂细胞周期的S期早期DNA复制的活跃区域富集,并且与全基因组背景相比显示出不同的DNA甲基化比率。超过一半的假定调控序列(通过其他方法鉴定)与本研究中检测到的CNS重叠。CNS中的变异与基因表达水平相关,CNS的缺失导致基因表达丧失。此外,在玉米亚基因组的背景下,CNS的进化与重复基因的功能多样化相关。我们的结果提供了对控制玉米CNS进化的分子过程的定量理解。