Bioanalytics, Institute of Biotechnology, Technische Universität Berlin, Berlin, Germany.
Data Analytics and Computational Statistics, Hasso Plattner Institute for Digital Engineering, Potsdam, Germany.
Nat Commun. 2021 May 28;12(1):3237. doi: 10.1038/s41467-021-23441-0.
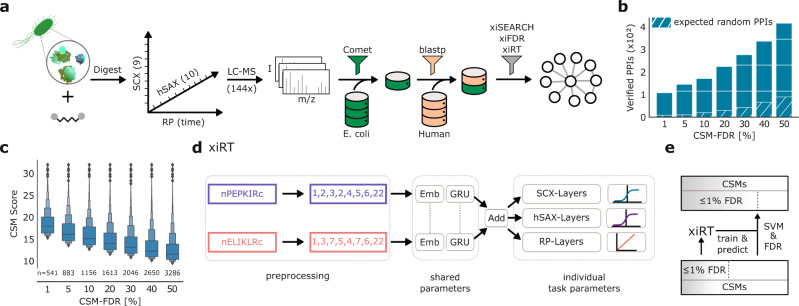
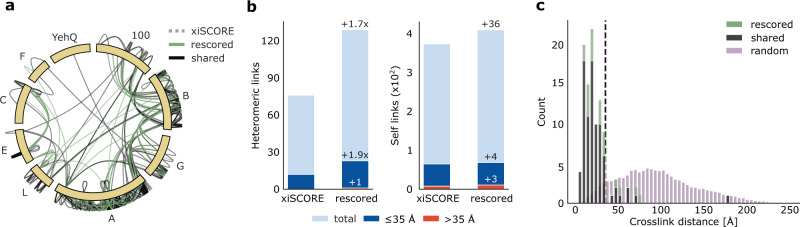
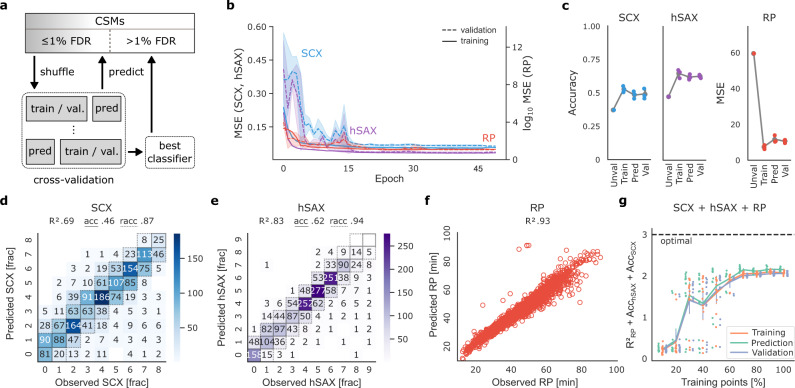
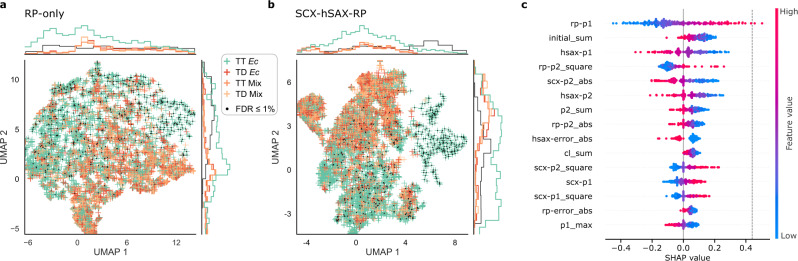
Crosslinking mass spectrometry has developed into a robust technique that is increasingly used to investigate the interactomes of organelles and cells. However, the incomplete and noisy information in the mass spectra of crosslinked peptides limits the numbers of protein-protein interactions that can be confidently identified. Here, we leverage chromatographic retention time information to aid the identification of crosslinked peptides from mass spectra. Our Siamese machine learning model xiRT achieves highly accurate retention time predictions of crosslinked peptides in a multi-dimensional separation of crosslinked E. coli lysate. Importantly, supplementing the search engine score with retention time features leads to a substantial increase in protein-protein interactions without affecting confidence. This approach is not limited to cell lysates and multi-dimensional separation but also improves considerably the analysis of crosslinked multiprotein complexes with a single chromatographic dimension. Retention times are a powerful complement to mass spectrometric information to increase the sensitivity of crosslinking mass spectrometry analyses.
交联质谱已发展成为一种强大的技术,越来越多地用于研究细胞器和细胞的相互作用组。然而,交联肽的质谱中不完整和嘈杂的信息限制了可以自信地识别的蛋白质-蛋白质相互作用的数量。在这里,我们利用色谱保留时间信息来帮助从质谱中鉴定交联肽。我们的暹罗机器学习模型 xiRT 实现了交联大肠杆菌裂解物多维分离中交联肽的高度准确保留时间预测。重要的是,将保留时间特征与搜索引擎得分相结合,可以在不影响置信度的情况下大大增加蛋白质-蛋白质相互作用。这种方法不仅限于细胞裂解物和多维分离,而且还极大地改善了具有单个色谱维度的交联多蛋白复合物的分析。保留时间是对质谱信息的有力补充,可提高交联质谱分析的灵敏度。