Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia, USA; Department of Biochemistry and Molecular Biology, University of Georgia, Athens, Georgia, USA.
Department of Biochemistry and Molecular Biology, University of Georgia, Athens, Georgia, USA.
J Biol Chem. 2021 Jul;297(1):100843. doi: 10.1016/j.jbc.2021.100843. Epub 2021 May 28.
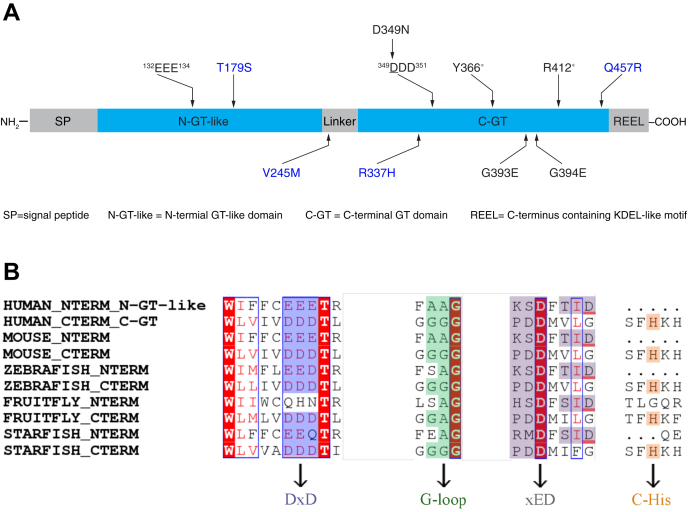
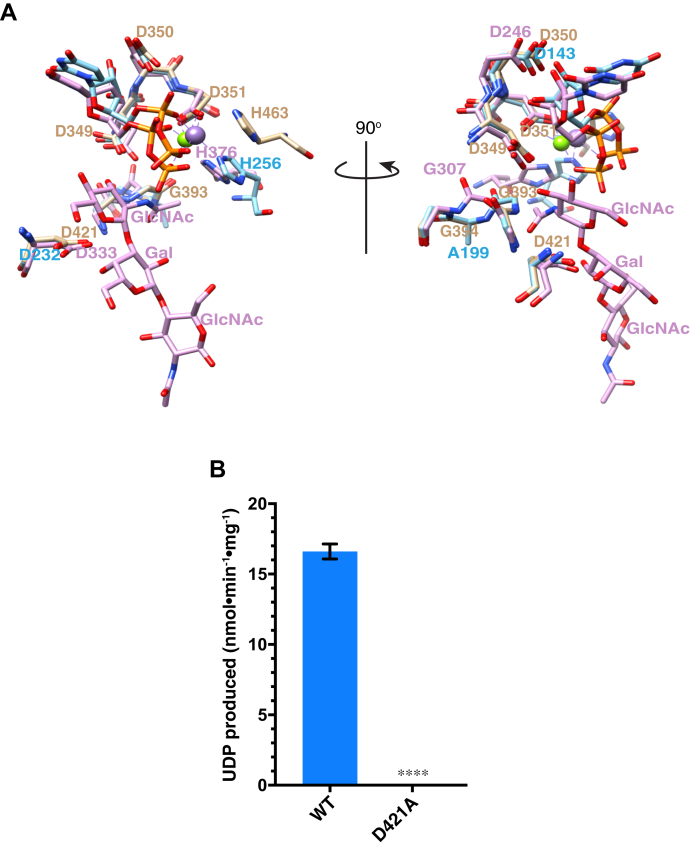
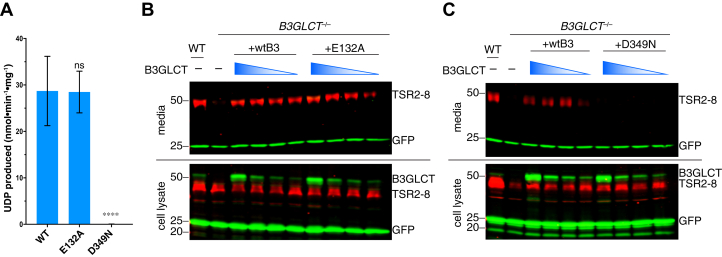
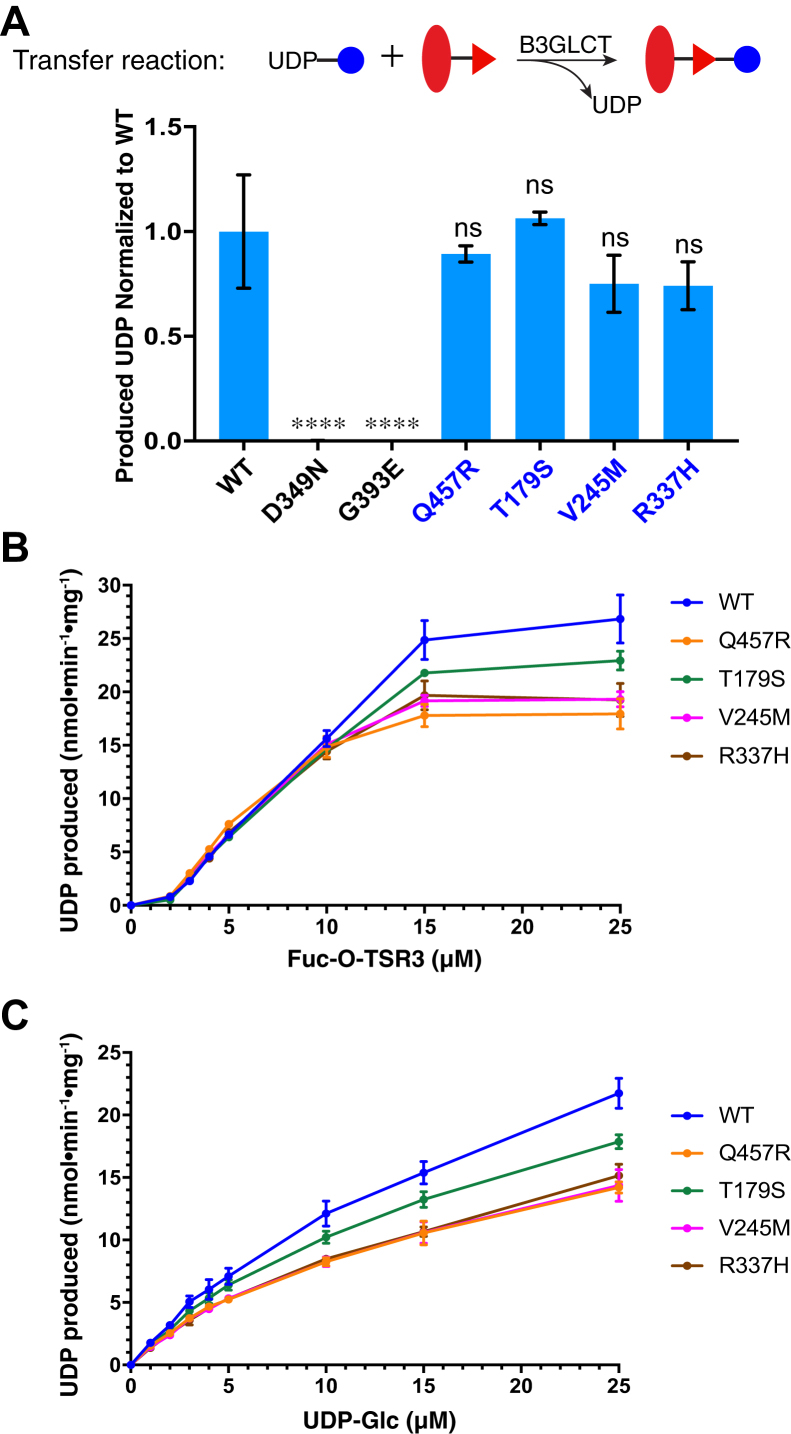
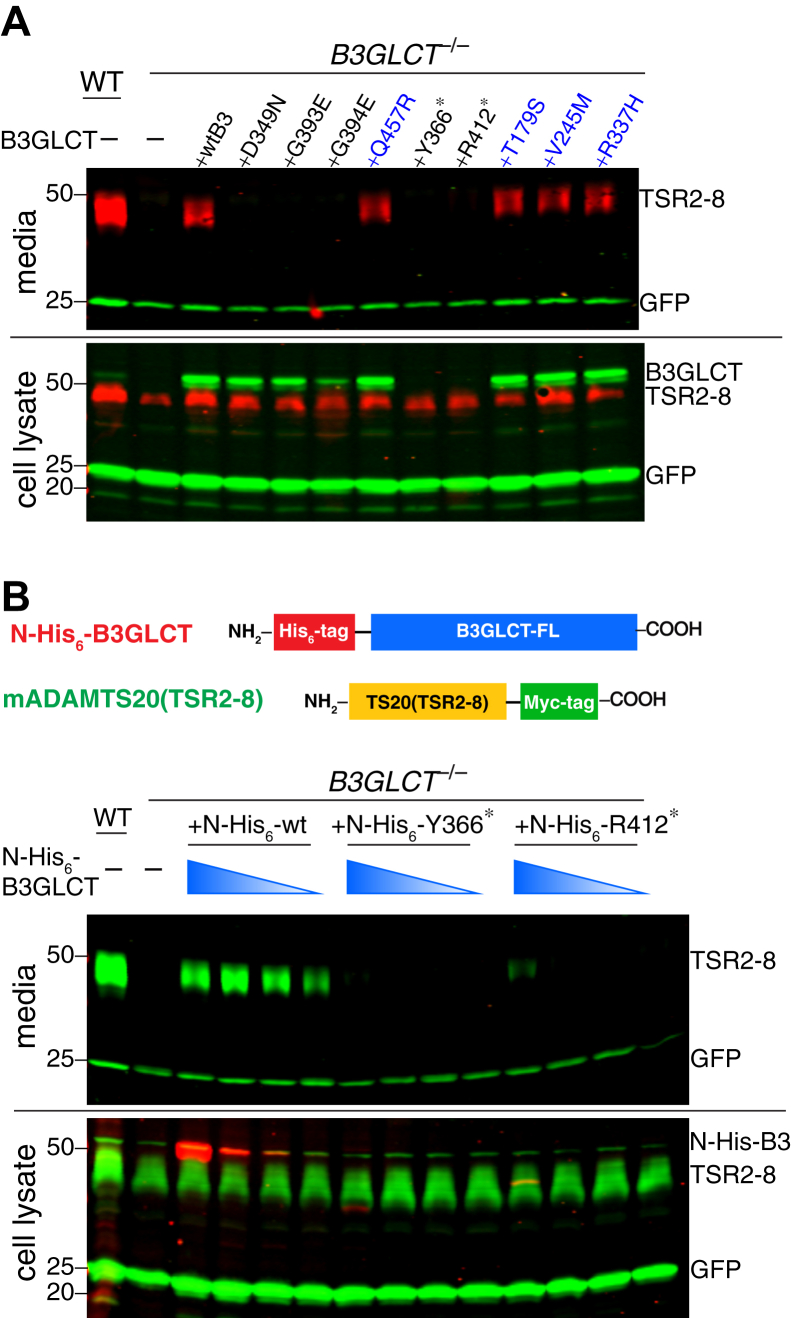
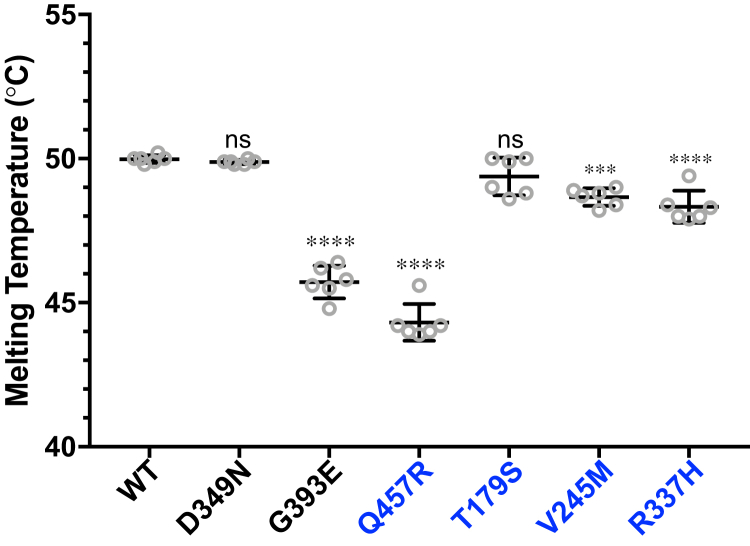
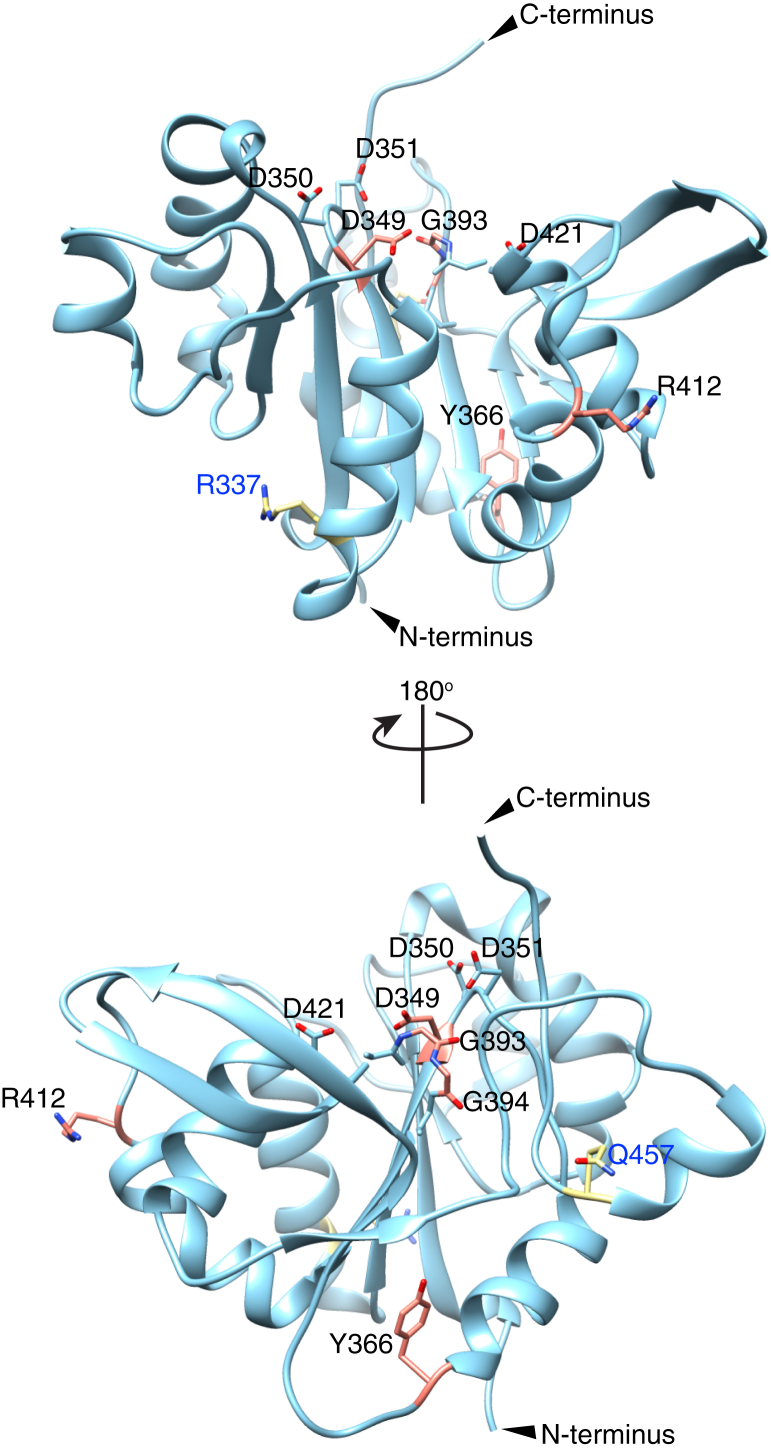
Peters Plus Syndrome (PTRPLS OMIM #261540) is a severe congenital disorder of glycosylation where patients have multiple structural anomalies, including Peters anomaly of the eye (anterior segment dysgenesis), disproportionate short stature, brachydactyly, dysmorphic facial features, developmental delay, and variable additional abnormalities. PTRPLS patients and some Peters Plus-like (PTRPLS-like) patients (who only have a subset of PTRPLS phenotypes) have mutations in the gene encoding β1,3-glucosyltransferase (B3GLCT). B3GLCT catalyzes the transfer of glucose to O-linked fucose on thrombospondin type-1 repeats. Most B3GLCT substrate proteins belong to the ADAMTS superfamily and play critical roles in extracellular matrix. We sought to determine whether the PTRPLS or PTRPLS-like mutations abrogated B3GLCT activity. B3GLCT has two putative active sites, one in the N-terminal region and the other in the C-terminal glycosyltransferase domain. Using sequence analysis and in vitro activity assays, we demonstrated that the C-terminal domain catalyzes transfer of glucose to O-linked fucose. We also generated a homology model of B3GLCT and identified D421 as the catalytic base. PTRPLS and PTRPLS-like mutations were individually introduced into B3GLCT, and the mutated enzymes were evaluated using in vitro enzyme assays and cell-based functional assays. Our results demonstrated that PTRPLS mutations caused loss of B3GLCT enzymatic activity and/or significantly reduced protein stability. In contrast, B3GLCT with PTRPLS-like mutations retained enzymatic activity, although some showed a minor destabilizing effect. Overall, our data supports the hypothesis that loss of glucose from B3GLCT substrate proteins is responsible for the defects observed in PTRPLS patients, but not for those observed in PTRPLS-like patients.
彼得斯-plus 综合征(Peters Plus Syndrome,PTRPLS OMIM #261540)是一种严重的糖基化先天性疾病,患者存在多种结构异常,包括眼部彼得斯异常(眼前节发育不良)、不成比例的身材矮小、短指畸形、面部畸形、发育迟缓以及其他可变的异常。PTRPLS 患者和一些彼得斯-plus 样(Peters Plus-like,PTRPLS-like)患者(仅具有 PTRPLS 表型的一部分)存在编码β1,3-葡糖基转移酶(B3GLCT)的基因突变。B3GLCT 催化葡萄糖向血小板反应蛋白-1 型重复物上的 O 连接岩藻糖的转移。大多数 B3GLCT 底物蛋白属于 ADAMTS 超家族,在细胞外基质中发挥关键作用。我们试图确定 PTRPLS 或 PTRPLS-like 突变是否使 B3GLCT 失活。B3GLCT 有两个假定的活性位点,一个位于 N 端区域,另一个位于 C 端糖基转移酶结构域。通过序列分析和体外活性测定,我们证明 C 端结构域催化葡萄糖向 O 连接岩藻糖的转移。我们还构建了 B3GLCT 的同源模型,并确定 D421 为催化碱。将 PTRPLS 和 PTRPLS-like 突变分别引入 B3GLCT,然后通过体外酶测定和基于细胞的功能测定评估突变酶。结果表明,PTRPLS 突变导致 B3GLCT 酶活性丧失和/或蛋白稳定性显著降低。相比之下,具有 PTRPLS-like 突变的 B3GLCT 保留了酶活性,尽管一些表现出轻微的不稳定作用。总体而言,我们的数据支持这样的假设,即 B3GLCT 底物蛋白上的葡萄糖缺失是 PTRPLS 患者缺陷的原因,但不是 PTRPLS-like 患者缺陷的原因。