Department of Cancer Biology, Mayo Clinic, 4500 San Pablo Road South, Jacksonville, FL 32224, USA.
Department of Microbiology and Plant Pathology, University of California, 900 University, Riverside, CA 92521, USA.
Biomolecules. 2021 May 23;11(6):787. doi: 10.3390/biom11060787.
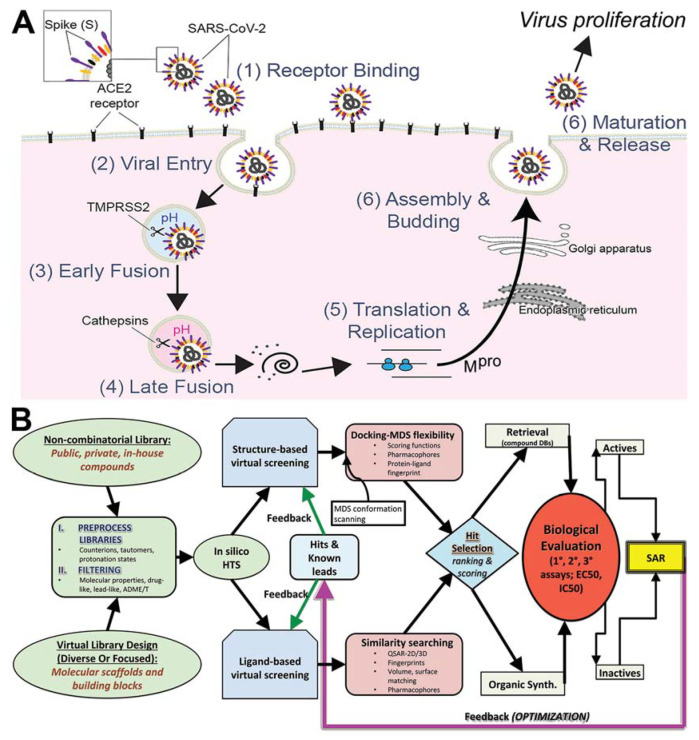
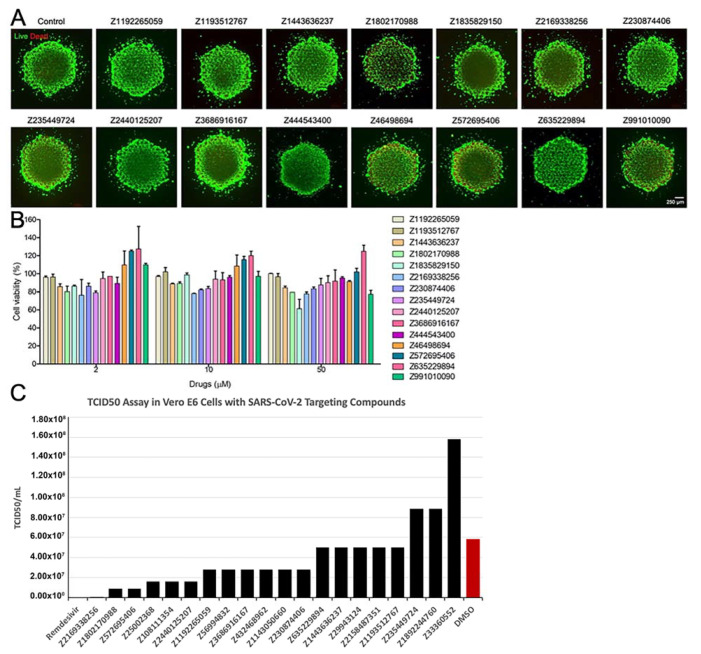
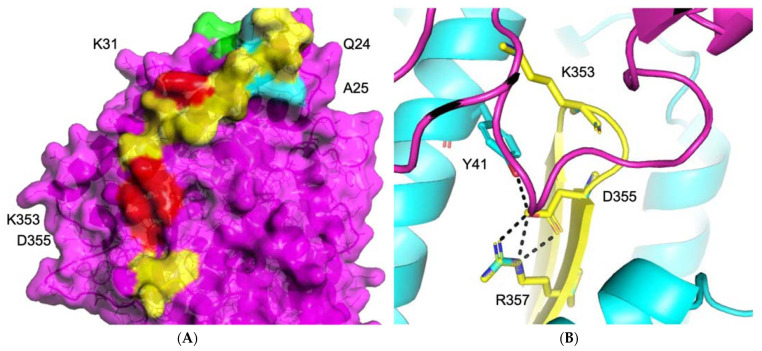
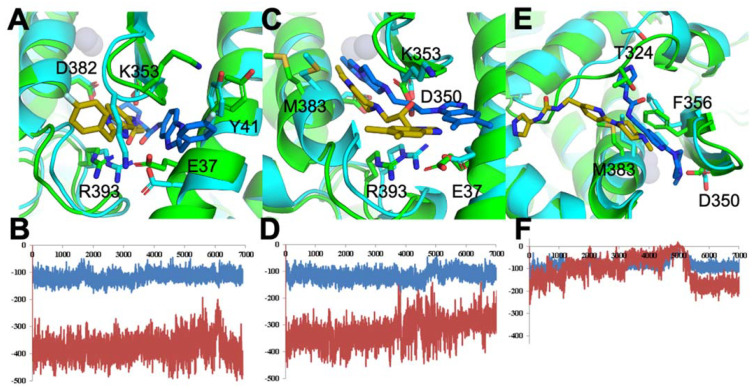
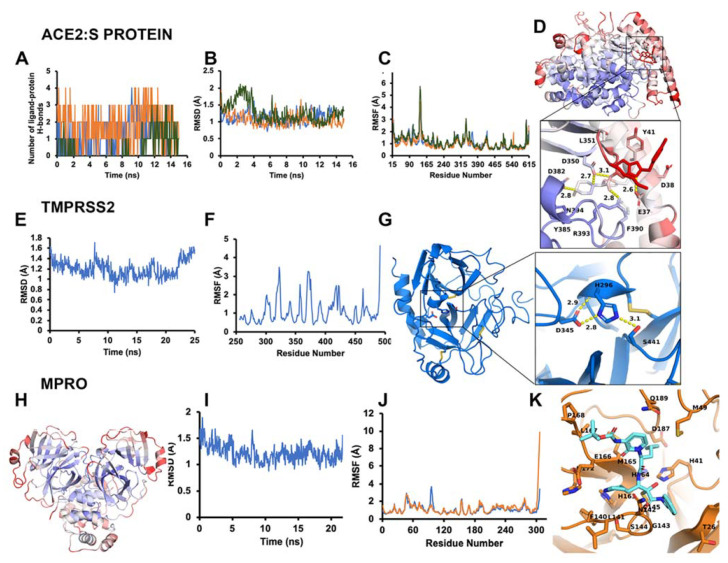
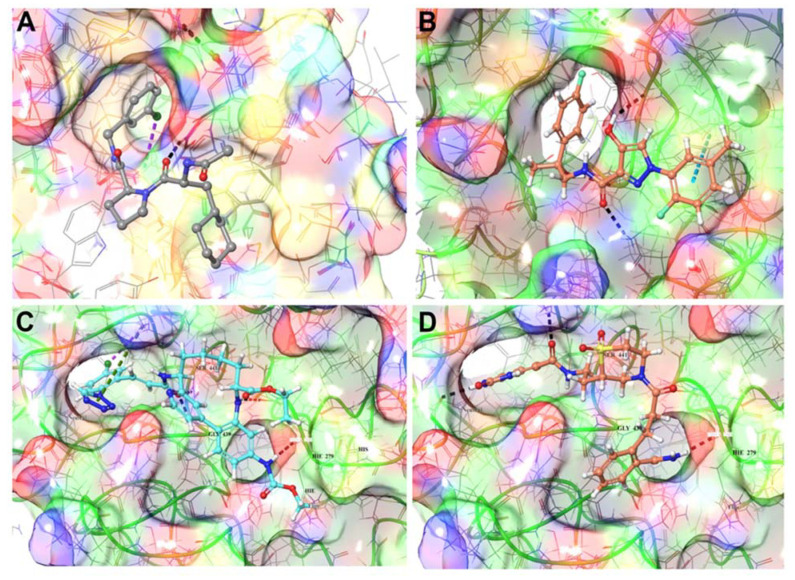
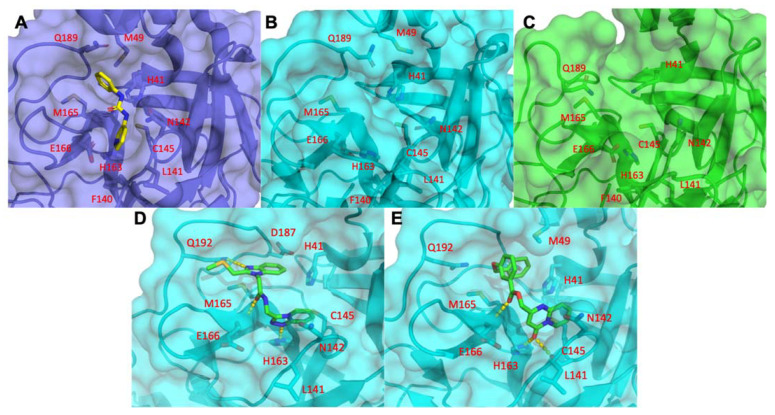
COVID-19 is a devastating respiratory and inflammatory illness caused by a new coronavirus that is rapidly spreading throughout the human population. Over the past 12 months, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for COVID-19, has already infected over 160 million (>20% located in United States) and killed more than 3.3 million people around the world (>20% deaths in USA). As we face one of the most challenging times in our recent history, there is an urgent need to identify drug candidates that can attack SARS-CoV-2 on multiple fronts. We have therefore initiated a computational dynamics drug pipeline using molecular modeling, structure simulation, docking and machine learning models to predict the inhibitory activity of several million compounds against two essential SARS-CoV-2 viral proteins and their host protein interactors-S/Ace2, Tmprss2, Cathepsins L and K, and Mpro-to prevent binding, membrane fusion and replication of the virus, respectively. All together, we generated an ensemble of structural conformations that increase high-quality docking outcomes to screen over >6 million compounds including all FDA-approved drugs, drugs under clinical trial (>3000) and an additional >30 million selected chemotypes from fragment libraries. Our results yielded an initial set of 350 high-value compounds from both new and FDA-approved compounds that can now be tested experimentally in appropriate biological model systems. We anticipate that our results will initiate screening campaigns and accelerate the discovery of COVID-19 treatments.
COVID-19 是一种由新型冠状病毒引起的破坏性呼吸道和炎症疾病,该病毒正在迅速在人群中传播。在过去的 12 个月中,引起 COVID-19 的严重急性呼吸系统综合征冠状病毒 2(SARS-CoV-2)已经感染了超过 1.6 亿人(位于美国的超过 20%),并导致全球超过 330 万人死亡(美国的超过 20%)。由于我们正面临着近代史上最具挑战性的时期之一,因此迫切需要确定能够从多个方面攻击 SARS-CoV-2 的药物候选物。因此,我们使用分子建模、结构模拟、对接和机器学习模型启动了一个计算动力学药物管道,以预测数百万种化合物对两种重要的 SARS-CoV-2 病毒蛋白及其宿主蛋白相互作用物-S/Ace2、Tmprss2、组织蛋白酶 L 和 K 以及 Mpro 的抑制活性,以分别防止病毒结合、膜融合和复制。总的来说,我们生成了一组结构构象,以提高高质量对接结果,从而筛选超过 600 万种化合物,包括所有 FDA 批准的药物、临床试验中的药物(超过 3000 种)以及来自片段库的另外 3000 多万种选定的化学型。我们的结果从新的和 FDA 批准的化合物中产生了一组最初的 350 种高价值化合物,现在可以在适当的生物学模型系统中进行实验测试。我们预计,我们的结果将启动筛选活动,并加速 COVID-19 治疗方法的发现。