Medical Research Council (MRC)/Uganda Virus Research Institute (UVRI) and London School of Hygiene and Tropical Medicine (LSHTM) Uganda Research Unit, Entebbe 256, Uganda.
Department of General Virology, Uganda Virus Research Institute, Entebbe 256, Uganda.
Viruses. 2021 May 24;13(6):970. doi: 10.3390/v13060970.
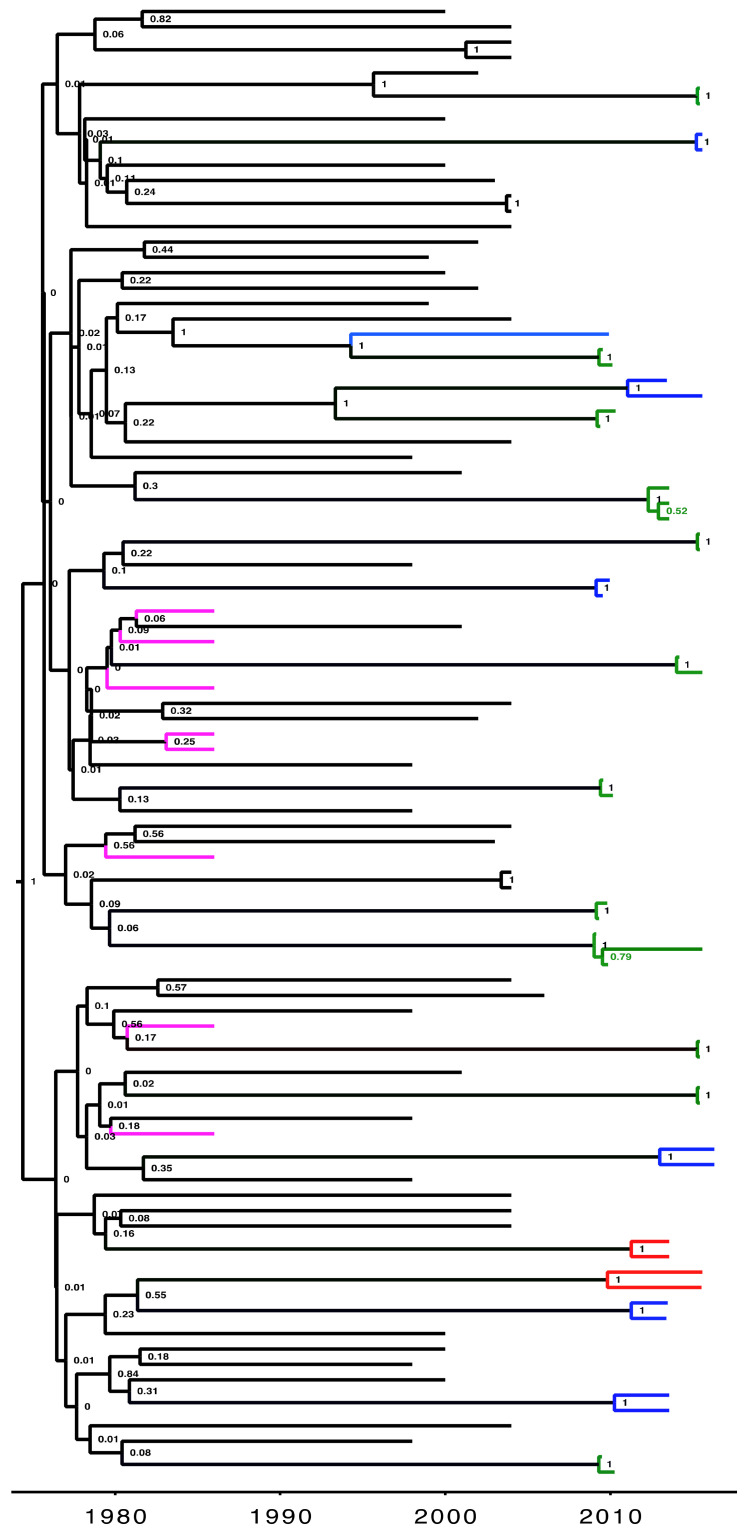
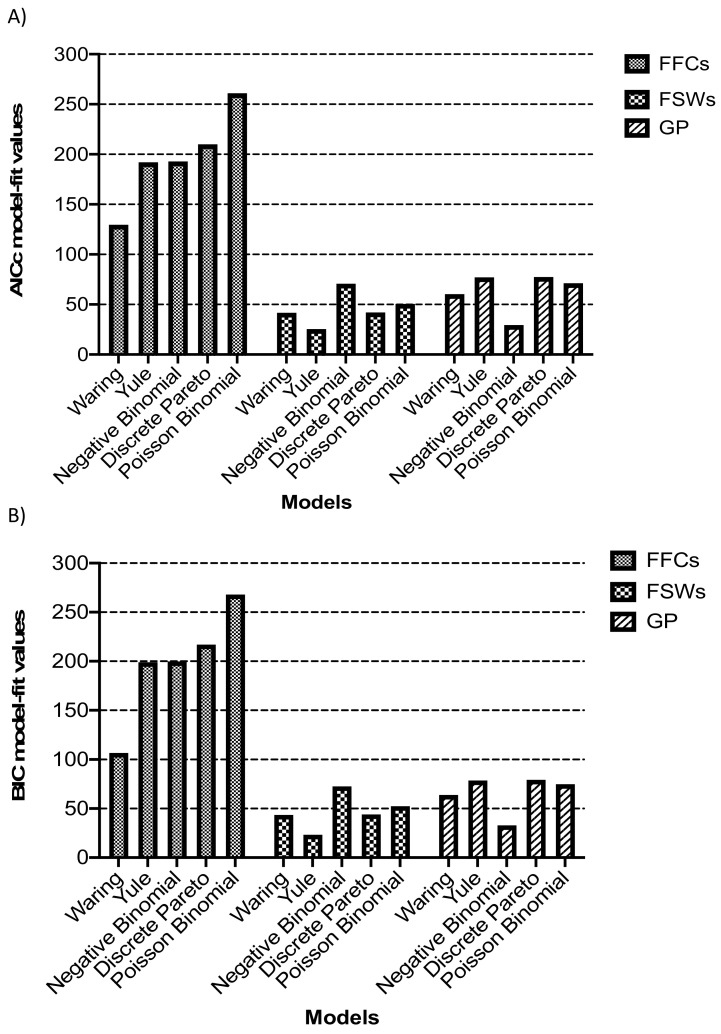
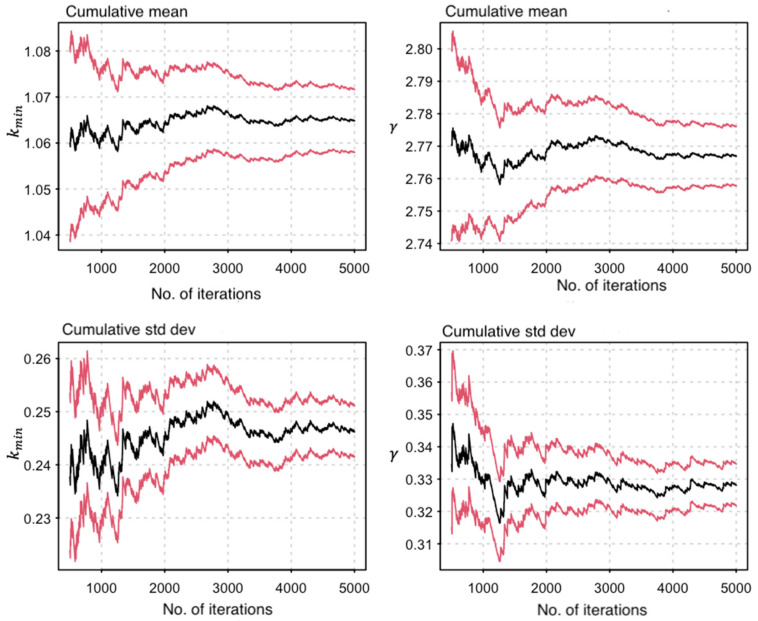
Phylogenetic inference is useful in characterising HIV transmission networks and assessing where prevention is likely to have the greatest impact. However, estimating parameters that influence the network structure is still scarce, but important in evaluating determinants of HIV spread. We analyzed 2017 HIV sequences (728 Lake Victoria fisherfolk communities (FFCs), 592 female sex workers (FSWs) and 697 general population (GP)) to identify transmission networks on Maximum Likelihood (ML) phylogenetic trees and refined them using time-resolved phylogenies. Network generative models were fitted to the observed degree distributions and network parameters, and corrected Akaike Information Criteria and Bayesian Information Criteria values were estimated. 347 (17.2%) HIV sequences were linked on ML trees (maximum genetic distance ≤4.5%, ≥95% bootstrap support) and, of these, 303 (86.7%) that consisted of pure A1 (n = 168) and D (n = 135) subtypes were analyzed in BEAST v1.8.4. The majority of networks (at least 40%) were found at a time depth of ≤5 years. The waring and yule models fitted best networks of FFCs and FSWs respectively while the negative binomial model fitted best networks in the GP. The network structure in the HIV-hyperendemic FFCs is likely to be scale-free and shaped by preferential attachment, in contrast to the GP. The findings support the targeting of interventions for FFCs in a timely manner for effective epidemic control. Interventions ought to be tailored according to the dynamics of the HIV epidemic in the target population and understanding the network structure is critical in ensuring the success of HIV prevention programs.
系统发育推断可用于描述 HIV 传播网络,并评估预防措施可能产生最大影响的地方。然而,估计影响网络结构的参数仍然很少,但对于评估 HIV 传播的决定因素很重要。我们分析了 2017 年 HIV 序列(728 个维多利亚湖渔民社区(FFC)、592 名性工作者(FSW)和 697 名普通人群(GP)),以在最大似然(ML)系统发育树上识别传播网络,并使用时间分辨系统发育树对其进行细化。将网络生成模型拟合到观察到的度数分布和网络参数上,并估计了校正的 Akaike 信息准则和贝叶斯信息准则值。347 个(17.2%)HIV 序列在 ML 树上存在关联(最大遗传距离≤4.5%,≥95%的自举支持),其中由纯 A1(n=168)和 D(n=135)亚型组成的 303 个(86.7%)进行了分析BEAST v1.8.4。大多数网络(至少 40%)的时间深度≤5 年。Warings 和 Yule 模型分别最适合 FFC 和 FSW 的网络,而负二项式模型最适合 GP 的网络。在 HIV 高流行的 FFC 中,网络结构可能是无标度的,并受到优先连接的影响,这与 GP 不同。这些发现支持及时针对 FFC 进行干预,以有效控制疫情。干预措施应根据目标人群中 HIV 疫情的动态进行调整,了解网络结构对于确保 HIV 预防计划的成功至关重要。