Jiang Zhengye, Shi Yanxi, Zhao Wenpeng, Zhang Yaya, Xie Yuanyuan, Zhang Bingchang, Tan Guowei, Wang Zhanxiang
Department of Neurosurgery, Xiamen Key Laboratory of Brain Center, The First Affiliated Hospital of Xiamen University, Xiamen, China.
School of Medicine, Institute of Neurosurgery, Xiamen University, Xiamen, China.
Front Neurol. 2021 May 19;12:610797. doi: 10.3389/fneur.2021.610797. eCollection 2021.
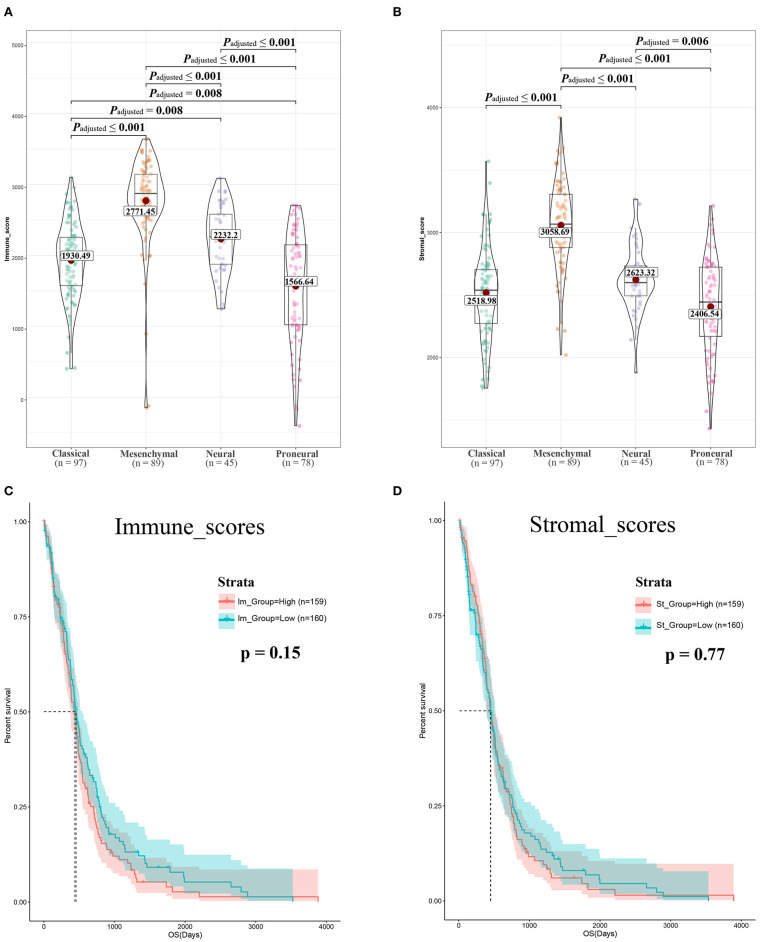
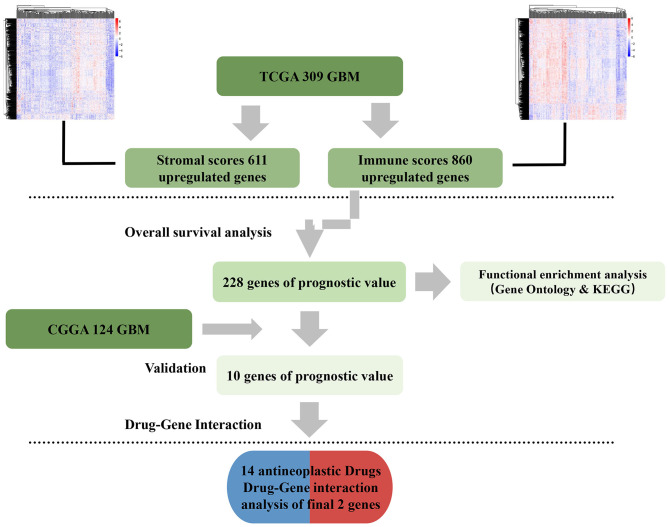
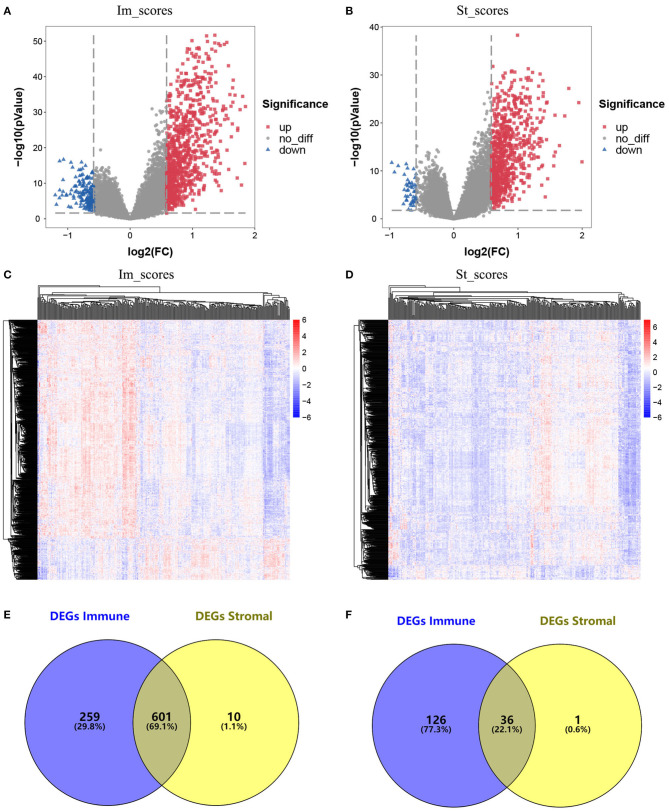
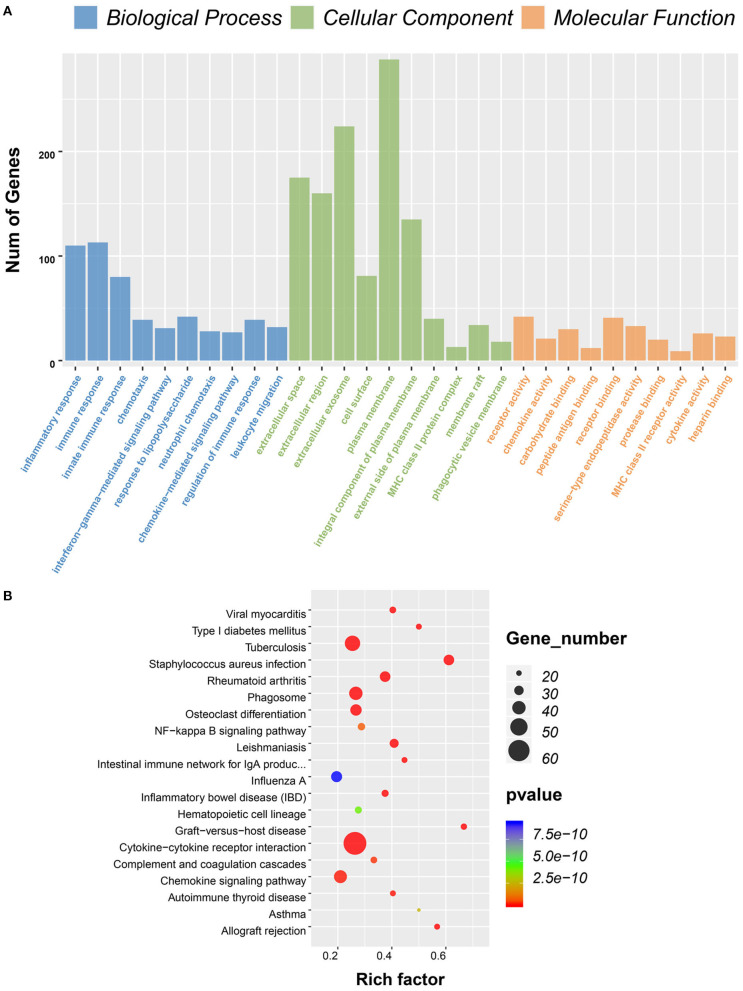
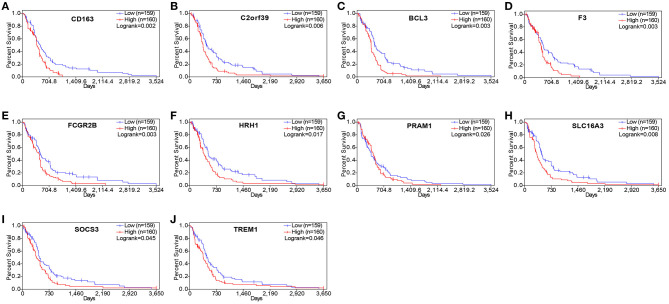
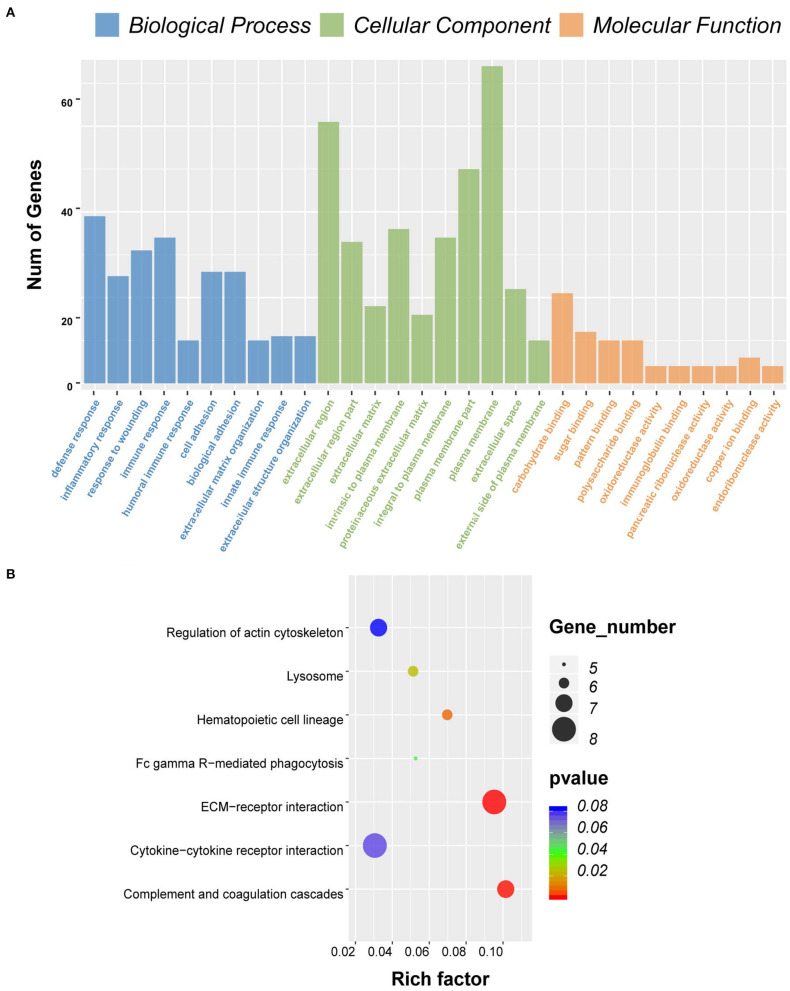
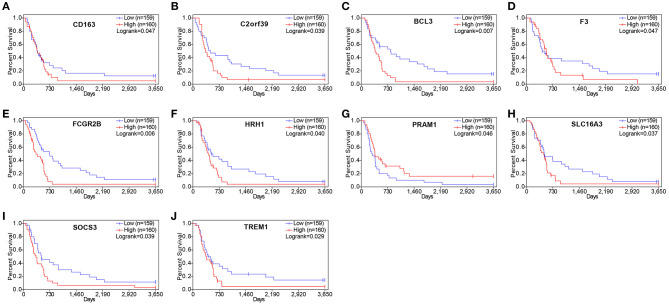
Although the tumor microenvironment (TME) is known to influence the prognosis of glioblastoma (GBM), the underlying mechanisms are not clear. This study aims to identify hub genes in the TME that affect the prognosis of GBM. The transcriptome profiles of the central nervous systems of GBM patients were downloaded from The Cancer Genome Atlas (TCGA). The ESTIMATE scoring algorithm was used to calculate immune and stromal scores. The application of these scores in histology classification was tested. Univariate Cox regression analysis was conducted to identify genes with prognostic value. Subsequently, functional enrichment analysis and protein-protein interaction (PPI) network analysis were performed to reveal the pathways and biological functions associated with the genes. Next, these prognosis genes were validated in an independent GBM cohort from the Chinese Glioma Genome Atlas (CGGA). Finally, the efficacy of current antitumor drugs targeting these genes against glioma was evaluated. Gene expression profiles and clinical data of 309 GBM samples were obtained from TCGA database. Higher immune and stromal scores were found to be significantly correlated with tissue type and poor overall survival (OS) ( = 0.15 and 0.77, respectively). Functional enrichment analysis identified 860 upregulated and 162 downregulated cross genes, which were mainly linked to immune response, inflammatory response, cell membrane, and receptor activity. Survival analysis identified 228 differentially expressed genes associated with the prognosis of GBM ( ≤ 0.05). A total of 48 hub genes were identified by the Cytoscape tool, and pathway enrichment analysis of the genes was performed using Database for Annotation, Visualization and Integrated Discovery (DAVID). The 228 genes were validated in an independent GBM cohort from the CGGA. In total, 10 genes were found to be significantly associated with prognosis of GBM. Finally, 14 antitumor drugs were identified by drug-gene interaction analysis. Here, 10 TME-related genes and 14 corresponding antitumor agents were found to be associated with the prognosis and OS of GBM.
虽然已知肿瘤微环境(TME)会影响胶质母细胞瘤(GBM)的预后,但其潜在机制尚不清楚。本研究旨在确定TME中影响GBM预后的关键基因。从癌症基因组图谱(TCGA)下载GBM患者中枢神经系统的转录组谱。使用ESTIMATE评分算法计算免疫和基质评分。测试了这些评分在组织学分类中的应用。进行单变量Cox回归分析以鉴定具有预后价值的基因。随后,进行功能富集分析和蛋白质-蛋白质相互作用(PPI)网络分析,以揭示与这些基因相关的途径和生物学功能。接下来,在中国胶质瘤基因组图谱(CGGA)的独立GBM队列中验证这些预后基因。最后,评估了目前针对这些基因的抗肿瘤药物对胶质瘤的疗效。从TCGA数据库获得了309个GBM样本的基因表达谱和临床数据。发现较高的免疫和基质评分与组织类型和较差的总生存期(OS)显著相关(分别为=0.15和0.77)。功能富集分析确定了860个上调和162个下调的交叉基因,这些基因主要与免疫反应、炎症反应、细胞膜和受体活性相关。生存分析确定了228个与GBM预后相关的差异表达基因(≤0.05)。通过Cytoscape工具共鉴定出48个关键基因,并使用注释、可视化和综合发现数据库(DAVID)对这些基因进行了途径富集分析。在来自CGGA的独立GBM队列中验证了这228个基因。总共发现10个基因与GBM的预后显著相关。最后,通过药物-基因相互作用分析鉴定了14种抗肿瘤药物。在这里,发现10个TME相关基因和14种相应的抗肿瘤药物与GBM的预后和OS相关。