Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, Canada.
Department of Medical Biophysics, University of Toronto, Toronto, Ontario, Canada.
Clin Cancer Res. 2021 Aug 1;27(15):4230-4244. doi: 10.1158/1078-0432.CCR-21-0110. Epub 2021 Jun 22.
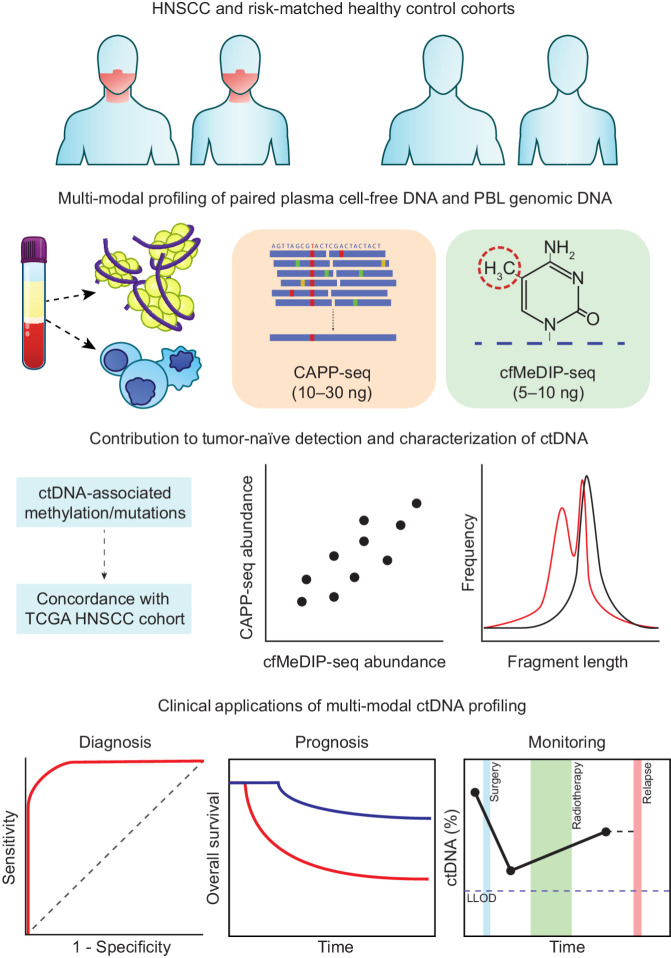
Circulating tumor DNA (ctDNA) enables personalized treatment strategies in oncology by providing a noninvasive source of clinical biomarkers. In patients with low ctDNA abundance, tumor-naïve methods are needed to facilitate clinical implementation. Here, using locoregionally confined head and neck squamous cell carcinoma (HNSCC) as an example, we demonstrate tumor-naïve detection of ctDNA by simultaneous profiling of mutations and methylation.
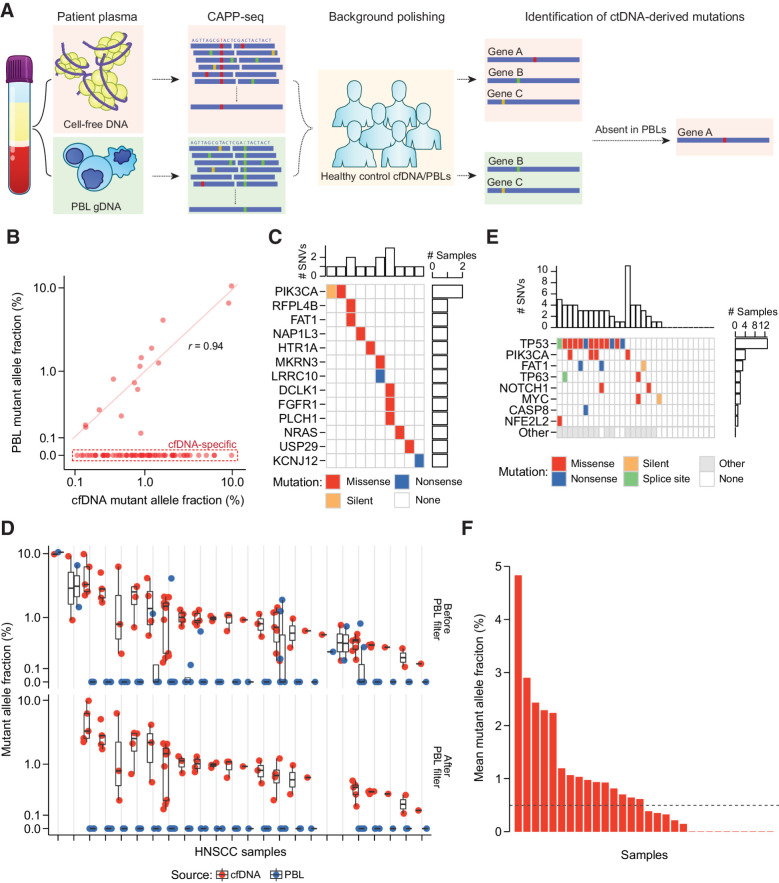
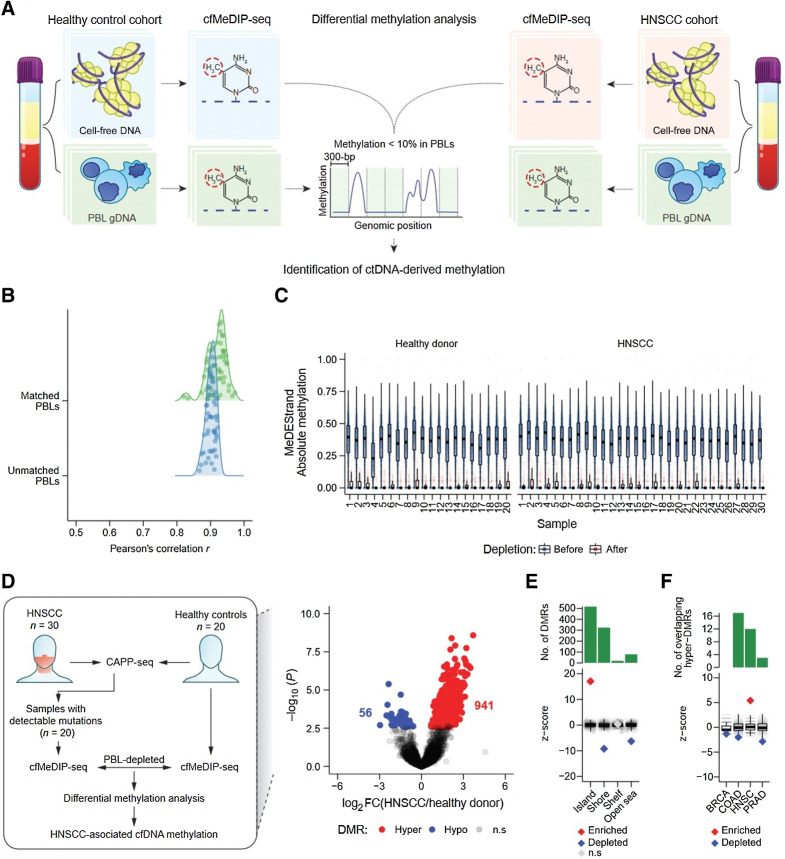
We conducted CAncer Personalized Profiling by deep Sequencing (CAPP-seq) and cell-free Methylated DNA ImmunoPrecipitation and high-throughput sequencing (cfMeDIP-seq) for detection of ctDNA-derived somatic mutations and aberrant methylation, respectively. We analyzed 77 plasma samples from 30 patients with stage I-IVA human papillomavirus-negative HNSCC as well as plasma samples from 20 risk-matched healthy controls. In addition, we analyzed leukocytes from patients and controls.
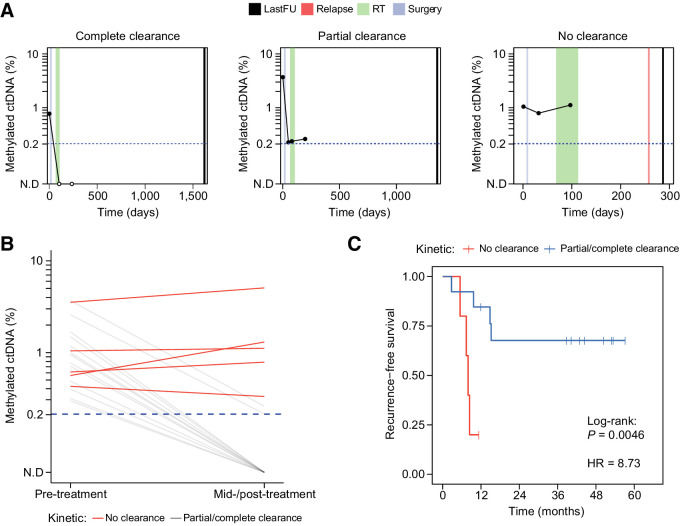
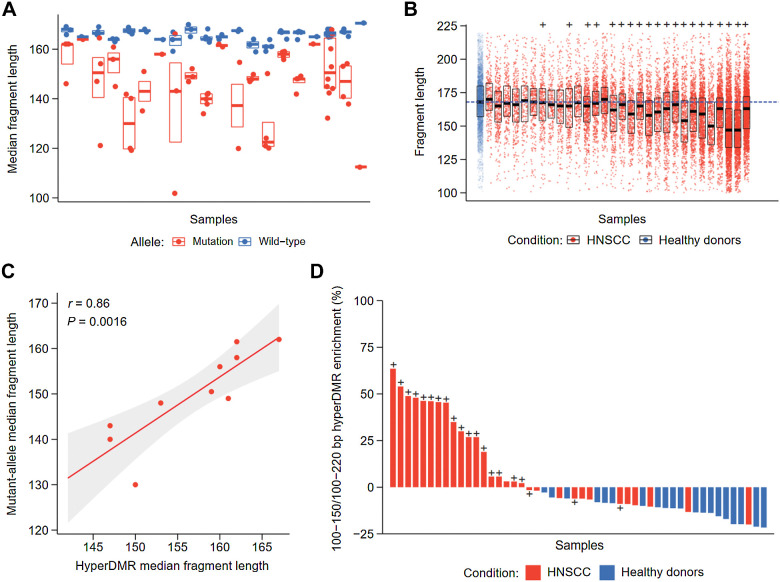
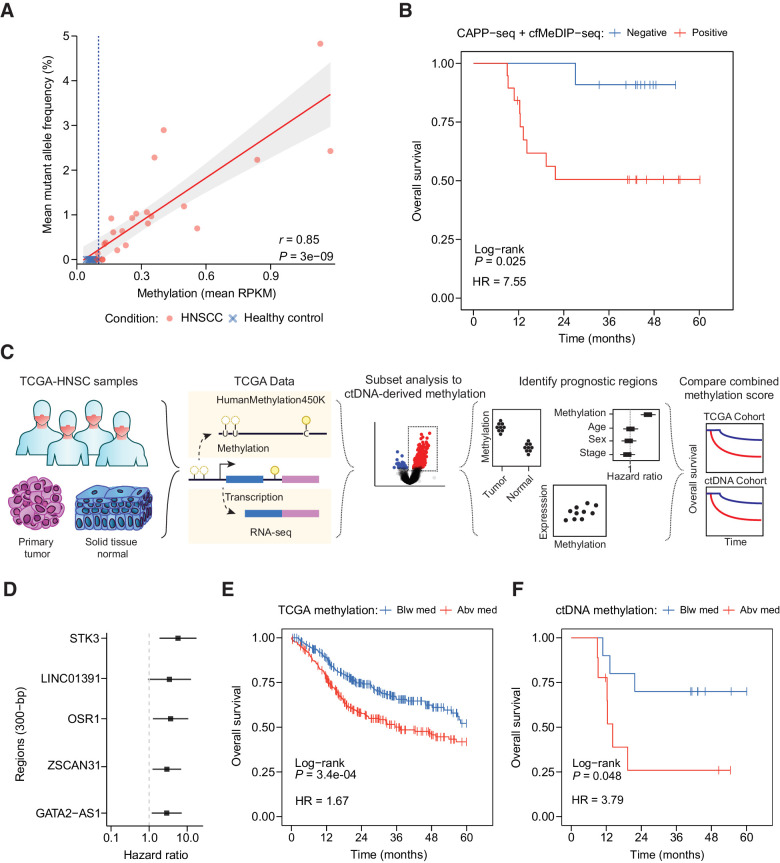
CAPP-seq identified mutations in 20 of 30 patients at frequencies similar to that of The Tumor Genome Atlas (TCGA). Differential methylation analysis of cfMeDIP-seq profiles identified 941 ctDNA-derived hypermethylated regions enriched for CpG islands and HNSCC-specific methylation patterns. Both methods demonstrated an association between ctDNA abundance and shorter fragment lengths. In addition, mutation- and methylation-based ctDNA abundance was highly correlated ( > 0.85). Patients with detectable pretreatment ctDNA by both methods demonstrated significantly worse overall survival (HR = 7.5; = 0.025) independent of clinical stage, with lack of ctDNA clearance post-treatment strongly correlating with recurrence. We further leveraged cfMeDIP-seq profiles to validate a prognostic signature identified from TCGA samples.
Tumor-naïve detection of ctDNA by multimodal profiling may facilitate biomarker discovery and clinical use in low ctDNA abundance applications.
循环肿瘤 DNA(ctDNA)通过提供非侵入性的临床生物标志物来源,为肿瘤学中的个性化治疗策略提供了可能。在 ctDNA 丰度较低的患者中,需要采用肿瘤原初方法来促进临床应用。在这里,我们以局部局限的头颈部鳞状细胞癌(HNSCC)为例,展示了通过突变和甲基化的同时分析来进行肿瘤原初 ctDNA 检测。
我们分别通过深度测序(CAPP-seq)和游离甲基化 DNA 免疫沉淀和高通量测序(cfMeDIP-seq)进行癌症个体化基因分析(CAncer Personalized Profiling by deep Sequencing,CAPP-seq)和细胞游离甲基化 DNA 免疫沉淀和高通量测序(cell-free Methylated DNA ImmunoPrecipitation and high-throughput sequencing,cfMeDIP-seq),以检测 ctDNA 衍生的体细胞突变和异常甲基化。我们分析了 30 例 I-IVA 期 HPV 阴性 HNSCC 患者的 77 个血浆样本以及 20 个风险匹配的健康对照者的血浆样本。此外,我们还分析了患者和对照者的白细胞。
CAPP-seq 在 30 例患者中的 20 例中以与肿瘤基因组图谱(TCGA)相似的频率鉴定出突变。cfMeDIP-seq 图谱的差异甲基化分析鉴定出 941 个 ctDNA 衍生的 hypermethylated 区,富含 CpG 岛和 HNSCC 特异性甲基化模式。两种方法均显示 ctDNA 丰度与较短片段长度之间存在关联。此外,基于突变和甲基化的 ctDNA 丰度高度相关(>0.85)。两种方法均能检测到预处理 ctDNA 的患者,其总体生存率显著降低(HR=7.5;=0.025),与治疗后 ctDNA 清除缺失强烈相关,且与复发密切相关。我们进一步利用 cfMeDIP-seq 图谱验证了从 TCGA 样本中鉴定出的预后签名。
通过多模式分析进行肿瘤原初 ctDNA 检测可能有助于发现生物标志物,并在 ctDNA 丰度较低的应用中促进临床应用。