Wang Quan, Wang Zhu, Zhang Zhen, Zhang Wei, Zhang Mengmeng, Shen Zhanlong, Ye Yingjiang, Jiang Kewei, Wang Shan
Department of Gastroenterological Surgery, Peking University People's Hospital, Beijing 100044, China.
Laboratory of Surgical Oncology, Beijing Key Laboratory of Colorectal Cancer Diagnosis and Treatment Research, Peking University People's Hospital, Beijing 100044, China.
Chin J Cancer Res. 2021 Apr 30;33(2):271-288. doi: 10.21147/j.issn.1000-9604.2021.02.13.
The goal of this study was to get preliminary insight on the intra-tumor heterogeneity in colitis-associated cancer (CAC) and to reveal a potential evolutionary trajectory from ulcerative colitis (UC) to CAC at the single-cell level.
Fresh samples of tumor tissues and adjacent UC tissues from a CAC patient with pT3N1M0 stage cancer were examined by single-cell RNA sequencing (scRNA-seq). Data from The Cancer Genome Atlas (TCGA) and The Human Protein Atlas were used to confirm the different expression levels in normal and tumor tissues and to determine their relationships with patient prognosis.
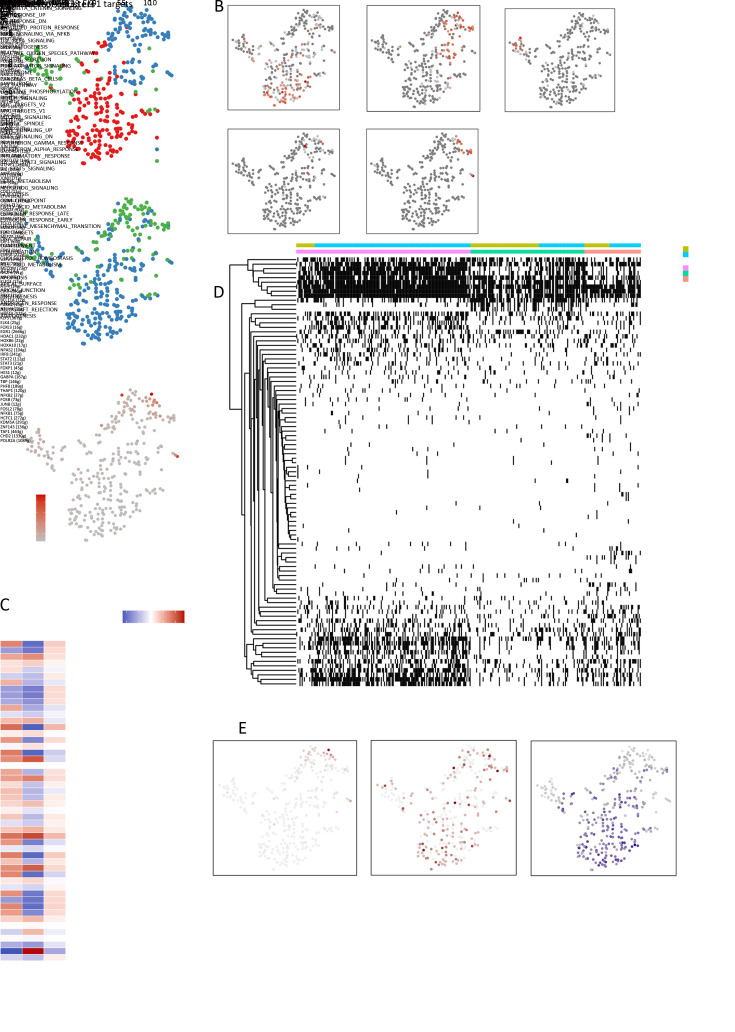
Ultimately, 4,777 single-cell transcriptomes (1,220 genes per cell) were examined, of which 2,250 (47%) and 2,527 (53%) originated from tumor and adjacent UC tissues, respectively. We defined the composition of cancer-associated stromal cells and identified six cell clusters, including myeloid, T and B cells, fibroblasts, endothelial and epithelial cells. Notable pathways and transcription factors involved in these cell clusters were analyzed and described. Moreover, the precise cellular composition and developmental trajectory from UC to UC-associated colon cancer were graphed, and it was predicted that , , and played a potential role in disease progression.
scRNA-seq technology revealed intra-tumor cell heterogeneity in UC-associated colon cancer, and might provide a promising direction to identify novel potential therapeutic targets in the evolution from UC to CAC.
本研究的目的是初步了解结肠炎相关癌(CAC)的肿瘤内异质性,并在单细胞水平揭示从溃疡性结肠炎(UC)到CAC的潜在进化轨迹。
对一名患有pT3N1M0期癌症的CAC患者的肿瘤组织和相邻UC组织的新鲜样本进行单细胞RNA测序(scRNA-seq)检查。来自癌症基因组图谱(TCGA)和人类蛋白质图谱的数据用于确认正常组织和肿瘤组织中的不同表达水平,并确定它们与患者预后的关系。
最终,共检测了4777个单细胞转录组(每个细胞1220个基因),其中2250个(47%)和2527个(53%)分别来自肿瘤组织和相邻的UC组织。我们定义了癌症相关基质细胞的组成,并确定了六个细胞簇,包括髓样细胞、T细胞和B细胞、成纤维细胞、内皮细胞和上皮细胞。对这些细胞簇中涉及的显著信号通路和转录因子进行了分析和描述。此外,绘制了从UC到UC相关结肠癌的精确细胞组成和发育轨迹,并预测 、 和 在疾病进展中发挥了潜在作用。
scRNA-seq技术揭示了UC相关结肠癌中的肿瘤内细胞异质性,并可能为识别从UC到CAC进化过程中的新型潜在治疗靶点提供一个有前景的方向。