Liu Jiaxin, Li Yimin, Gan Yaqi, Xiao Qing, Tian Ruotong, Shu Guang, Yin Gang
Department of Pathology, Xiangya Hospital, School of Basic Medical Sciences, Central South University, Changsha, China.
School of Basic Medical Sciences, Central South University, Changsha, China.
Front Cell Dev Biol. 2021 Jun 11;9:671211. doi: 10.3389/fcell.2021.671211. eCollection 2021.
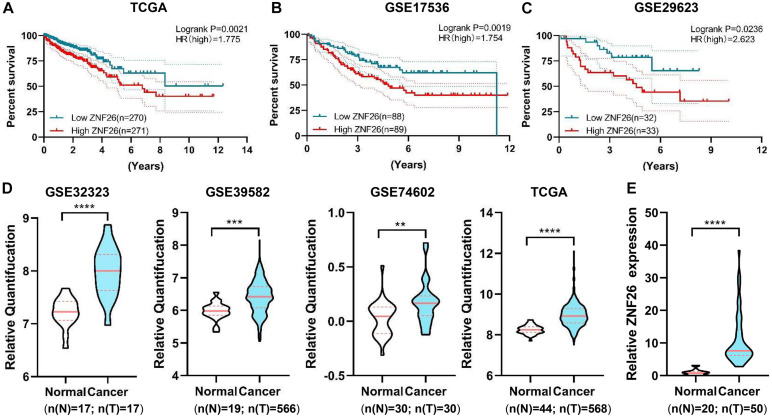
The dysregulation of transcriptional factors (TFs) leads to malignant growth and the development of colorectal cancer (CRC). Herein, we sought to identify the transcription factors relevant to the prognosis of colorectal cancer patients. We found 526 differentially expressed TFs using the TCGA database of colorectal cancer patients ( = 544) for the differential analysis of TFs ( = 1,665) with 210 upregulated genes as well as 316 downregulated genes. Subsequently, GO analysis and KEGG pathway analysis were performed for these differential genes for investigating their pathways and function. At the same time, we established a genetic risk scoring model for predicting the overall survival (OS) by using the mRNA expression levels of these differentially regulated TFs, and defined the CRC into low and high-risk categories which showed significant survival differences. The genetic risk scoring model included four high-risk genes (, , , and ) and two low-risk genes ( and ), and validated the OS in two GEO databases ( = 0.0023 for the GSE17536, = 0.0193 for the GSE29623). To analyze the genetic and epigenetic changes of these six risk-related TFs, a unified bioinformatics analysis was conducted. Among them, is progressive in CRC and its high expression is linked with a poor diagnosis as well. Knockdown of inhibits the proliferative capacity of CRC cells. Moreover, the positive association between and cyclins (CDK2, CCNE2, CDK6, CHEK1) was also identified. Therefore, as a novel biomarker, may be a promising candidate in the diagnosis and prognostic evaluation of colorectal cancer.
转录因子(TFs)的失调会导致恶性生长和结直肠癌(CRC)的发展。在此,我们试图鉴定与结直肠癌患者预后相关的转录因子。我们使用结直肠癌患者的TCGA数据库(n = 544)对TFs(n = 1,665)进行差异分析,发现了526个差异表达的TFs,其中210个基因上调,316个基因下调。随后,对这些差异基因进行了GO分析和KEGG通路分析,以研究它们的通路和功能。同时,我们利用这些差异调节的TFs的mRNA表达水平建立了一个遗传风险评分模型来预测总生存期(OS),并将CRC分为低风险和高风险类别,这两类显示出显著的生存差异。遗传风险评分模型包括四个高风险基因(、、、和)和两个低风险基因(和),并在两个GEO数据库中验证了OS(GSE17536的P = 0.0023,GSE29623的P = 0.0193)。为了分析这六个与风险相关的TFs的遗传和表观遗传变化,进行了统一的生物信息学分析。其中,在CRC中呈进展性,其高表达也与不良诊断相关。敲低可抑制CRC细胞的增殖能力。此外,还发现与细胞周期蛋白(CDK2、CCNE2、CDK6、CHEK1)之间存在正相关。因此,作为一种新型生物标志物,可能是结直肠癌诊断和预后评估中有前景的候选者。