Department of Statistical Sciences, University of Toronto, Toronto, Ontario, Canada.
Biostatistics Division, Dalla Lana School of Public Health, University of Toronto, Toronto, Ontario, Canada.
Genet Epidemiol. 2021 Oct;45(7):694-709. doi: 10.1002/gepi.22422. Epub 2021 Jul 5.
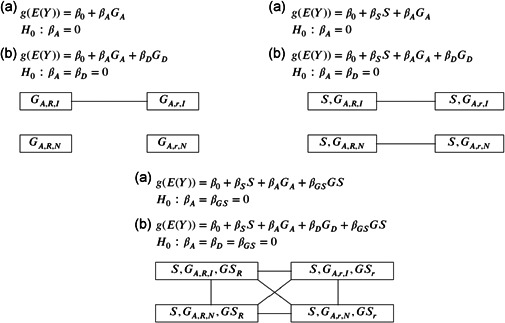
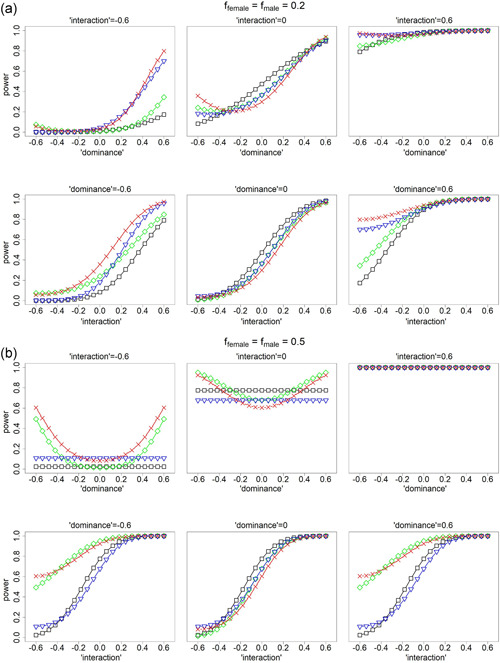
The X-chromosome is often excluded from genome-wide association studies because of analytical challenges. Some of the problems, such as the random, skewed, or no X-inactivation model uncertainty, have been investigated. Other considerations have received little to no attention, such as the value in considering nonadditive and gene-sex interaction effects, and the inferential consequence of choosing different baseline alleles (i.e., the reference vs. the alternative allele). Here we propose a unified and flexible regression-based association test for X-chromosomal variants. We provide theoretical justifications for its robustness in the presence of various model uncertainties, as well as for its improved power when compared with the existing approaches under certain scenarios. For completeness, we also revisit the autosomes and show that the proposed framework leads to a more robust approach than the standard method. Finally, we provide supporting evidence by revisiting several published association studies. Supporting Information for this article are available online.
X 染色体通常会从全基因组关联研究中排除,因为存在分析挑战。一些问题,如随机、偏斜或不存在 X 失活模型不确定性,已经得到了研究。其他一些因素则很少受到关注,例如考虑非加性和基因性别相互作用效应的价值,以及选择不同基线等位基因(即参考等位基因与替代等位基因)的推断后果。在这里,我们提出了一种统一而灵活的基于回归的 X 染色体变异关联检验方法。我们为其在存在各种模型不确定性时的稳健性以及在某些情况下与现有方法相比的改进功效提供了理论依据。为了完整性,我们还重新研究了常染色体,并表明所提出的框架比标准方法更稳健。最后,我们通过重新研究几个已发表的关联研究提供了支持证据。本文的支持信息可在线获取。