Computational Structural Biology Group, Bijvoet Centre for Biomolecular Research, Faculty of Science-Chemistry, Utrecht University, Padualaan 8, 3584 CH Utrecht, the Netherlands.
Physics Department, University of Cagliari, Cittadella Universitaria, S.P. 8 km 0.700, 09042 Monserrato, Italy.
J Chem Theory Comput. 2021 Sep 14;17(9):5944-5954. doi: 10.1021/acs.jctc.1c00336. Epub 2021 Aug 3.
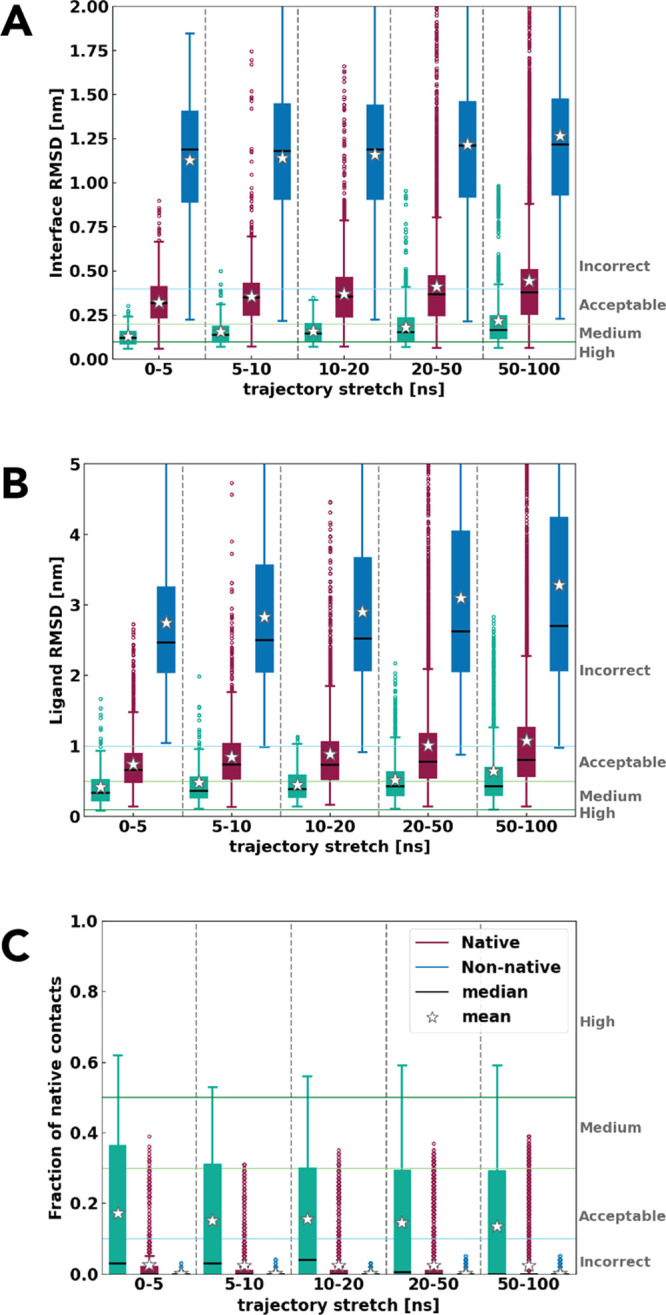
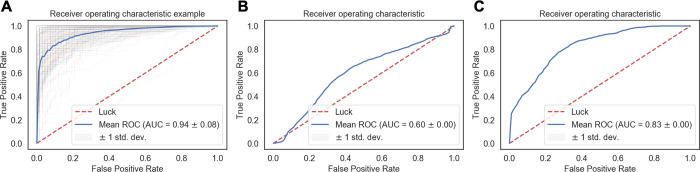
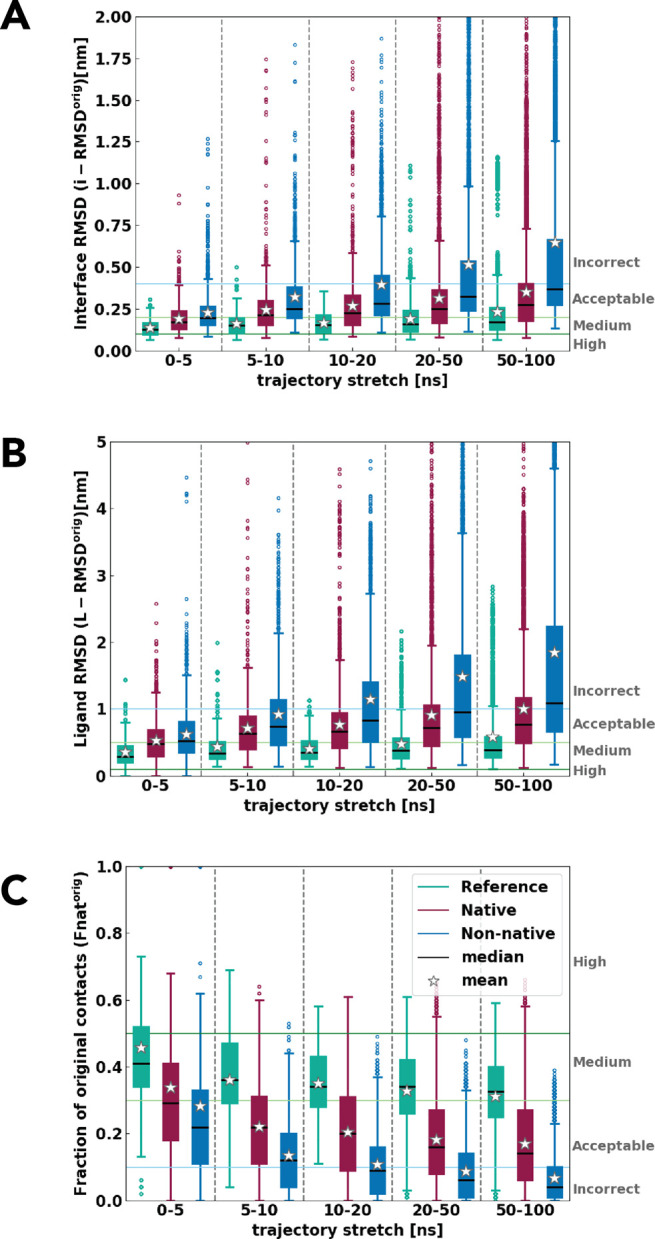
Molecular docking excels at creating a plethora of potential models of protein-protein complexes. To correctly distinguish the favorable, native-like models from the remaining ones remains, however, a challenge. We assessed here if a protocol based on molecular dynamics (MD) simulations would allow distinguishing native from non-native models to complement scoring functions used in docking. To this end, the first models for 25 protein-protein complexes were generated using HADDOCK. Next, MD simulations complemented with machine learning were used to discriminate between native and non-native complexes based on a combination of metrics reporting on the stability of the initial models. Native models showed higher stability in almost all measured properties, including the key ones used for scoring in the Critical Assessment of PRedicted Interaction (CAPRI) competition, namely the positional root mean square deviations and fraction of native contacts from the initial docked model. A random forest classifier was trained, reaching a 0.85 accuracy in correctly distinguishing native from non-native complexes. Reasonably modest simulation lengths of the order of 50-100 ns are sufficient to reach this accuracy, which makes this approach applicable in practice.
分子对接擅长生成大量蛋白质-蛋白质复合物的潜在模型。然而,正确区分有利的、类似天然的模型与其余模型仍然是一个挑战。我们在这里评估了一种基于分子动力学 (MD) 模拟的方案是否可以区分天然和非天然模型,以补充对接中使用的评分函数。为此,使用 HADDOCK 为 25 个蛋白质-蛋白质复合物生成了第一批模型。接下来,使用机器学习补充的 MD 模拟根据报告初始模型稳定性的组合指标来区分天然和非天然复合物。天然模型在几乎所有测量的特性中表现出更高的稳定性,包括在关键评估预测相互作用 (Critical Assessment of PRedicted Interaction,CAPRI) 竞赛中用于评分的特性,即位置均方根偏差和初始对接模型中天然接触的分数。随机森林分类器进行了训练,在正确区分天然和非天然复合物方面的准确率达到 0.85。达到这个准确率所需的适度模拟长度为 50-100 ns 左右,这使得这种方法在实践中具有可操作性。