Department of Electronics and Information Engineering, Jeonbuk National University, Jeonju 54896, Republic of Korea.
Department of Electrical Engineering, Telkom University, Bandung 40257, West Java, Indonesia.
Int J Mol Sci. 2024 May 29;25(11):5957. doi: 10.3390/ijms25115957.
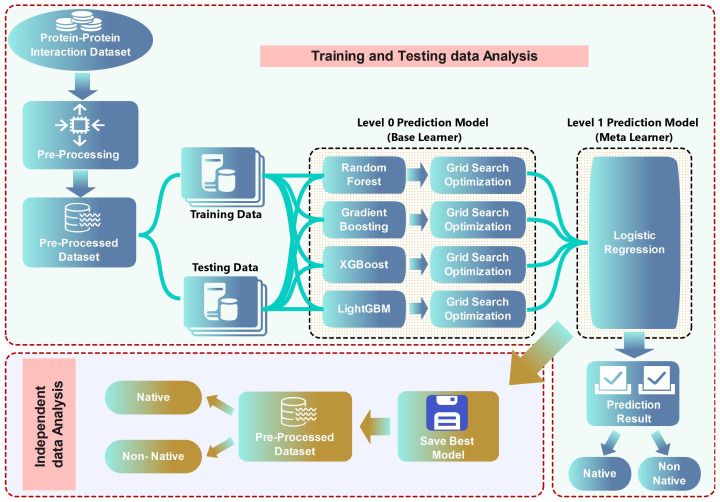
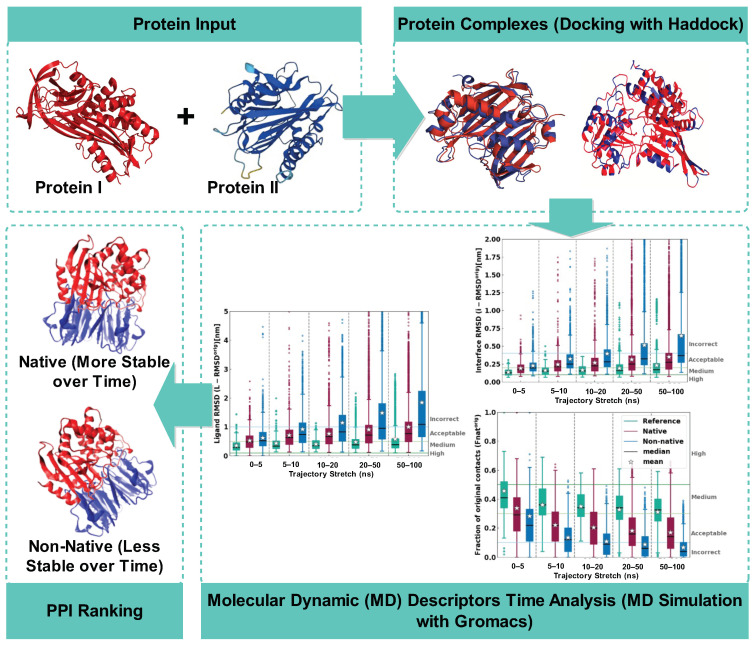
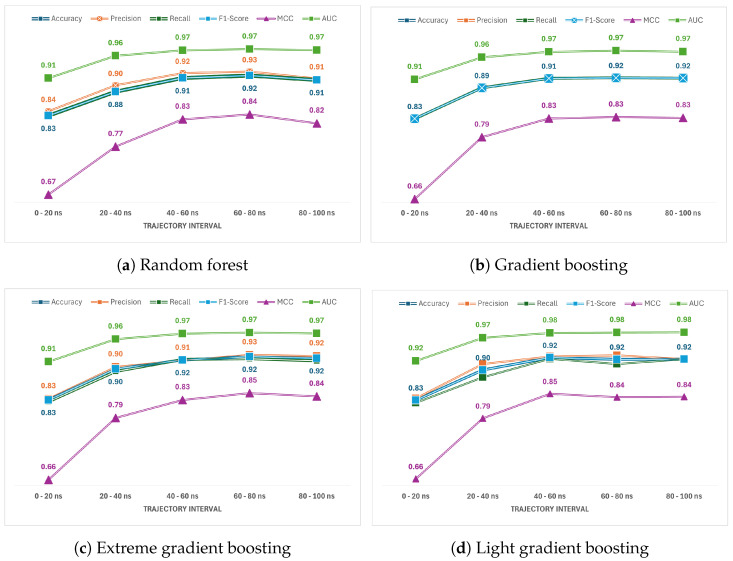
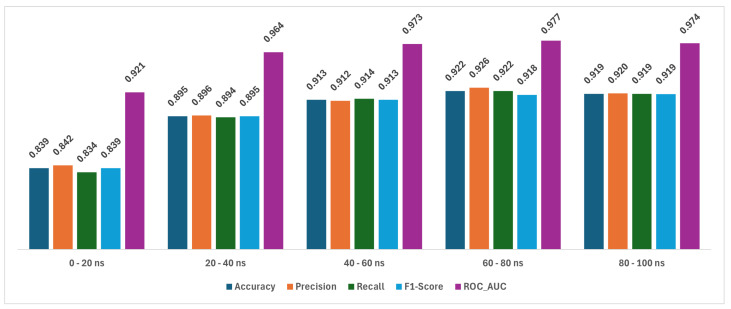
In this study, we present an innovative approach to improve the prediction of protein-protein interactions (PPIs) through the utilization of an ensemble classifier, specifically focusing on distinguishing between native and non-native interactions. Leveraging the strengths of various base models, including random forest, gradient boosting, extreme gradient boosting, and light gradient boosting, our ensemble classifier integrates these diverse predictions using a logistic regression meta-classifier. Our model was evaluated using a comprehensive dataset generated from molecular dynamics simulations. While the gains in AUC and other metrics might seem modest, they contribute to a model that is more robust, consistent, and adaptable. To assess the effectiveness of various approaches, we compared the performance of logistic regression to four baseline models. Our results indicate that logistic regression consistently underperforms across all evaluated metrics. This suggests that it may not be well-suited to capture the complex relationships within this dataset. Tree-based models, on the other hand, appear to be more effective for problems involving molecular dynamics simulations. Extreme gradient boosting (XGBoost) and light gradient boosting (LightGBM) are optimized for performance and speed, handling datasets effectively and incorporating regularizations to avoid over-fitting. Our findings indicate that the ensemble method enhances the predictive capability of PPIs, offering a promising tool for computational biology and drug discovery by accurately identifying potential interaction sites and facilitating the understanding of complex protein functions within biological systems.
在这项研究中,我们提出了一种创新的方法,通过利用集成分类器来提高蛋白质-蛋白质相互作用(PPIs)的预测能力,特别是专注于区分天然和非天然相互作用。利用各种基础模型的优势,包括随机森林、梯度提升、极端梯度提升和轻梯度提升,我们的集成分类器使用逻辑回归元分类器整合了这些不同的预测。我们的模型使用从分子动力学模拟生成的综合数据集进行了评估。虽然 AUC 和其他指标的增益看起来微不足道,但它们有助于构建更稳健、一致和适应性强的模型。为了评估各种方法的有效性,我们将逻辑回归的性能与四个基线模型进行了比较。我们的结果表明,逻辑回归在所有评估指标上的表现都不一致。这表明它可能不适合捕捉这个数据集内部的复杂关系。另一方面,基于树的模型对于涉及分子动力学模拟的问题似乎更有效。极端梯度提升(XGBoost)和轻梯度提升(LightGBM)是为了性能和速度而优化的,能够有效地处理数据集,并包含正则化以避免过拟合。我们的研究结果表明,集成方法增强了 PPIs 的预测能力,通过准确识别潜在的相互作用位点,并促进对生物系统中复杂蛋白质功能的理解,为计算生物学和药物发现提供了一种有前途的工具。