College of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, Korea.
Int J Mol Sci. 2020 Sep 1;21(17):6339. doi: 10.3390/ijms21176339.
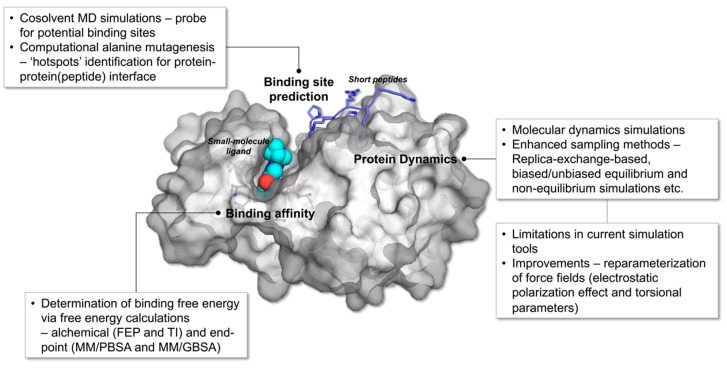
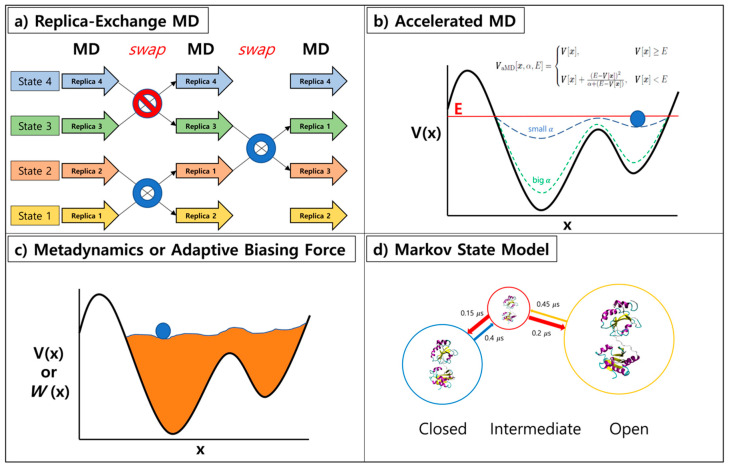
Molecular dynamics (MD) simulation is a rigorous theoretical tool that when used efficiently could provide reliable answers to questions pertaining to the structure-function relationship of proteins. Data collated from protein dynamics can be translated into useful statistics that can be exploited to sieve thermodynamics and kinetics crucial for the elucidation of mechanisms responsible for the modulation of biological processes such as protein-ligand binding and protein-protein association. Continuous modernization of simulation tools enables accurate prediction and characterization of the aforementioned mechanisms and these qualities are highly beneficial for the expedition of drug development when effectively applied to structure-based drug design (SBDD). In this review, current all-atom MD simulation methods, with focus on enhanced sampling techniques, utilized to examine protein structure, dynamics, and functions are discussed. This review will pivot around computer calculations of protein-ligand and protein-protein systems with applications to SBDD. In addition, we will also be highlighting limitations faced by current simulation tools as well as the improvements that have been made to ameliorate their efficiency.
分子动力学(MD)模拟是一种严格的理论工具,如果使用得当,可以为与蛋白质的结构-功能关系相关的问题提供可靠的答案。从蛋白质动力学中收集的数据可以转化为有用的统计数据,这些数据可以用于筛选对于阐明负责调节生物过程(如蛋白质-配体结合和蛋白质-蛋白质相互作用)的机制至关重要的热力学和动力学。模拟工具的不断现代化使得能够准确预测和表征上述机制,当有效地应用于基于结构的药物设计(SBDD)时,这些特性对于药物开发的加速非常有益。在这篇综述中,讨论了当前用于研究蛋白质结构、动力学和功能的全原子 MD 模拟方法,重点是增强采样技术。这篇综述将围绕蛋白质-配体和蛋白质-蛋白质系统的计算机计算展开,并将其应用于 SBDD。此外,我们还将强调当前模拟工具所面临的局限性,以及为提高其效率而做出的改进。