Neuroscience Graduate Program, Kavli Institute for Fundamental Neuroscience, Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, USA.
Department of Neurology, Kavli Institute for Fundamental Neuroscience, Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, USA.
Cell Rep. 2021 Aug 3;36(5):109483. doi: 10.1016/j.celrep.2021.109483.
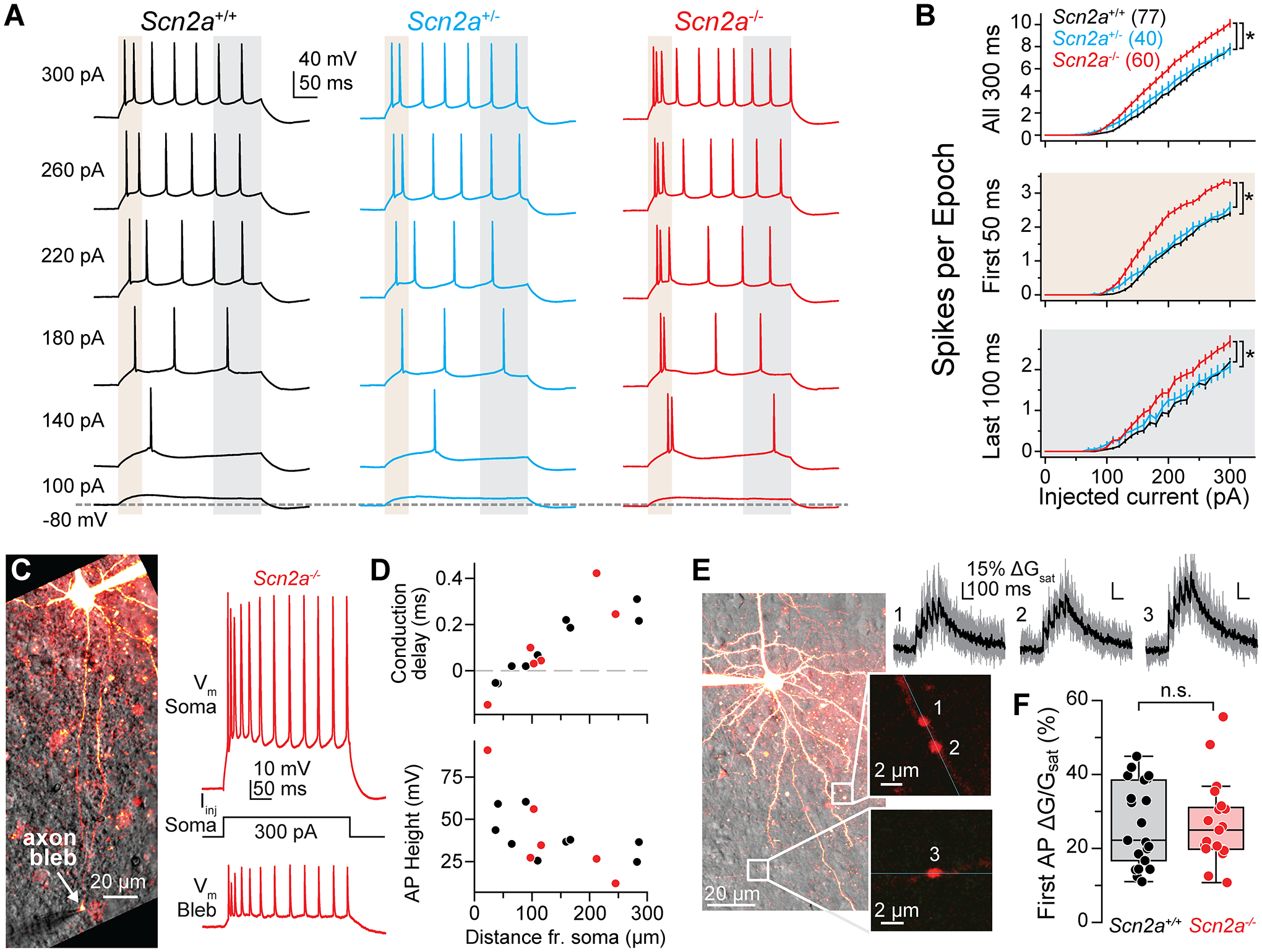
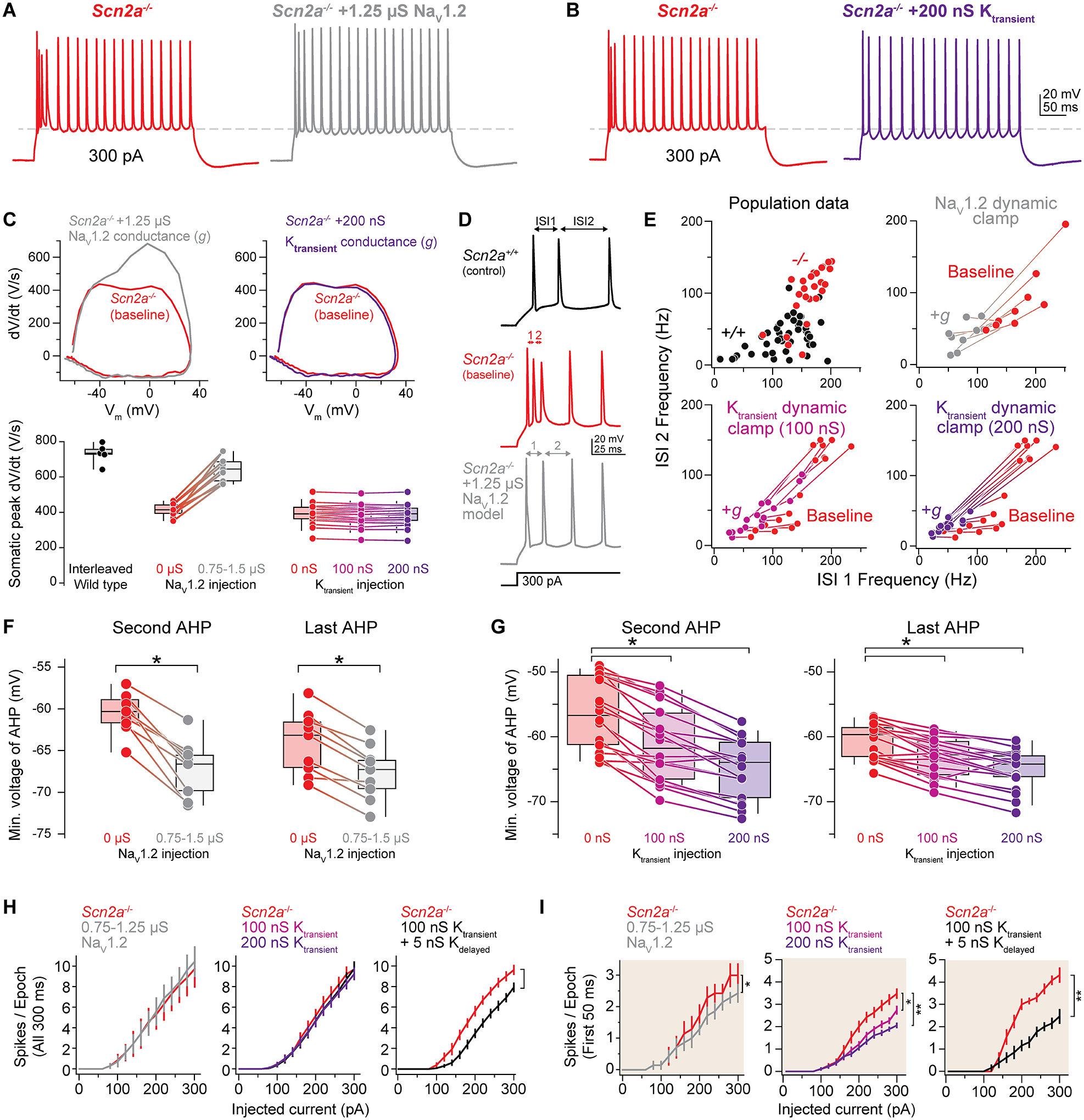
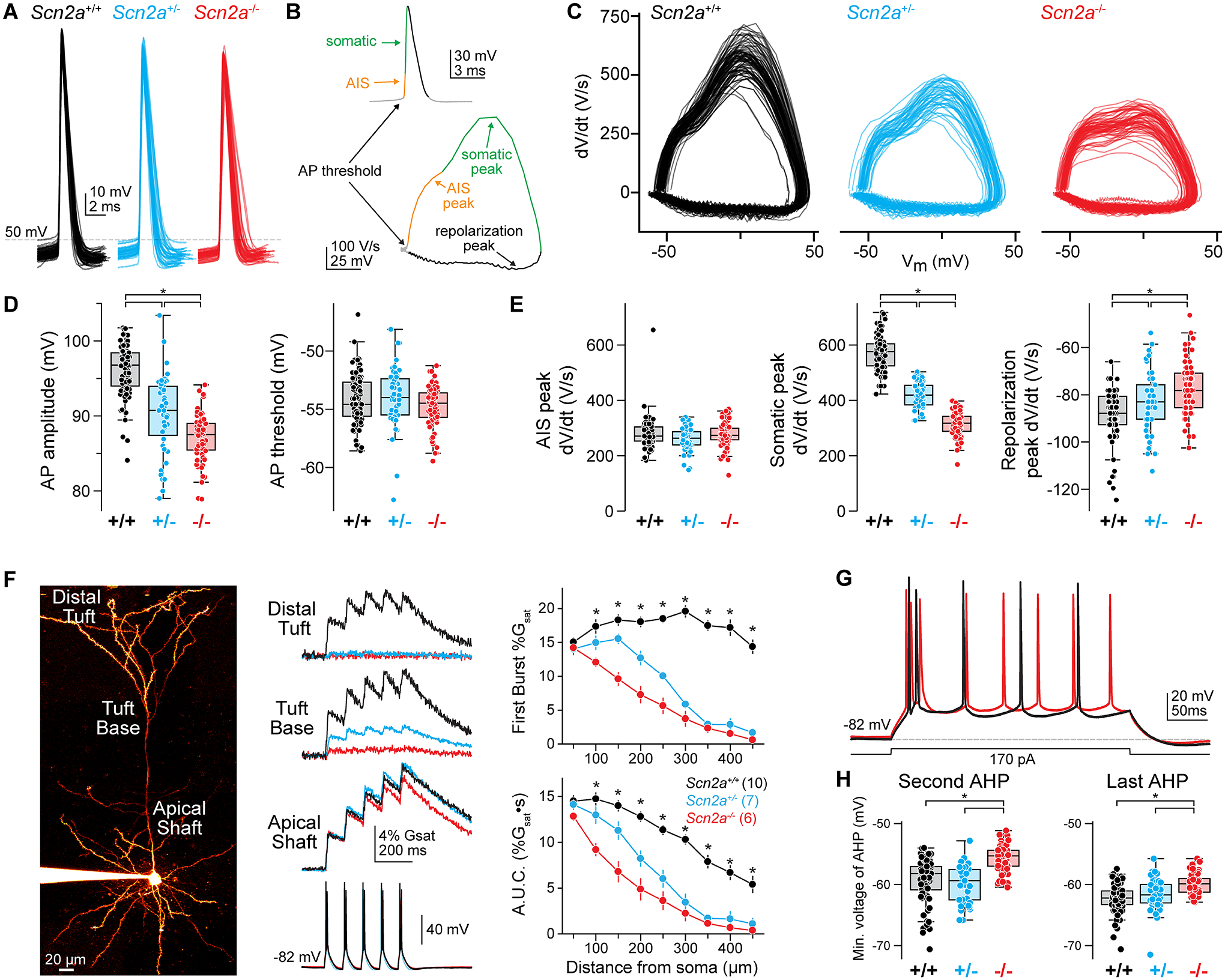
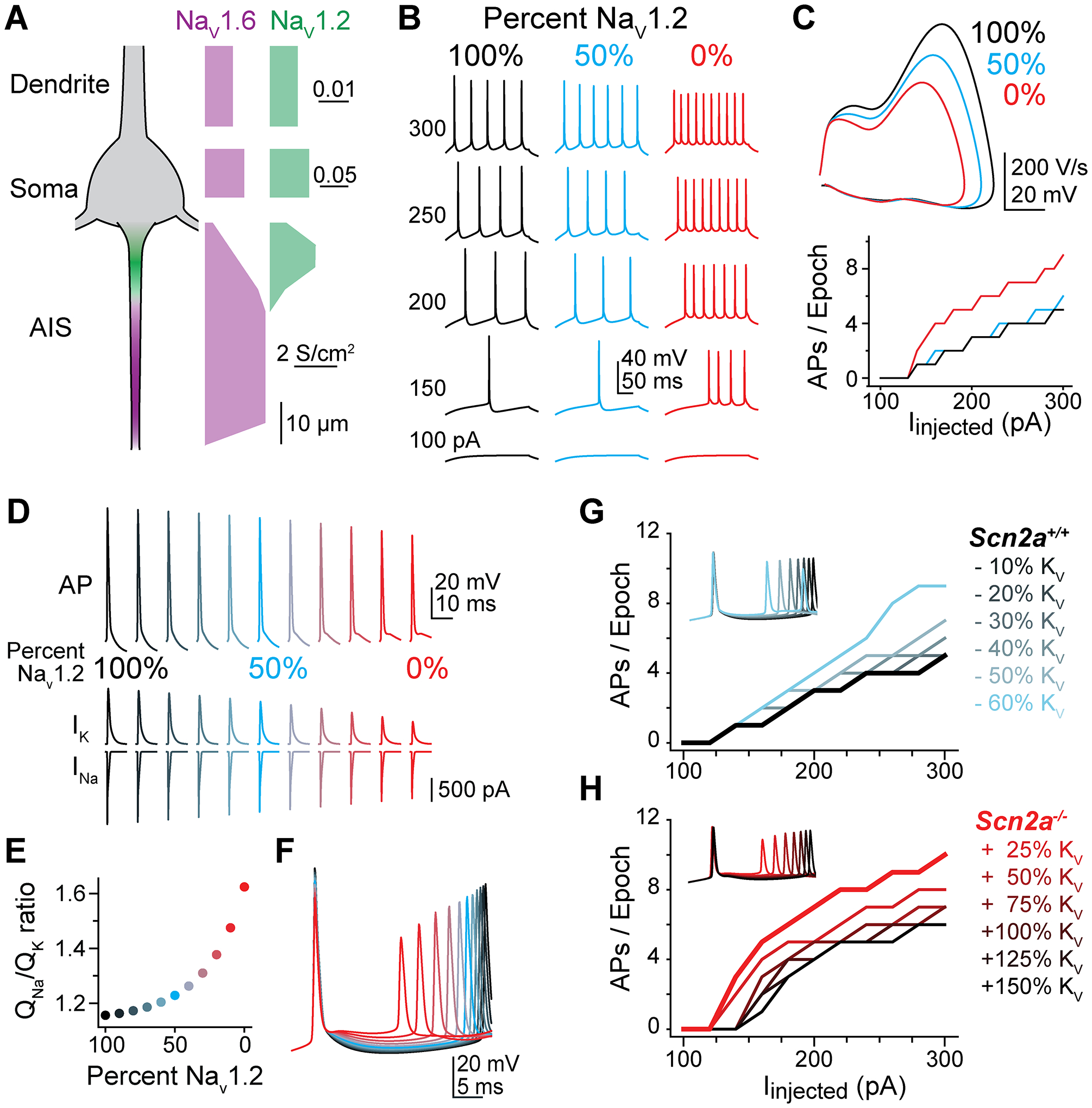
Loss-of-function variants in the gene SCN2A, which encodes the sodium channel Na1.2, are strongly associated with autism spectrum disorder and intellectual disability. An estimated 20%-30% of children with these variants also suffer from epilepsy, with altered neuronal activity originating in neocortex, a region where Na1.2 channels are expressed predominantly in excitatory pyramidal cells. This is paradoxical, as sodium channel loss in excitatory cells would be expected to dampen neocortical activity rather than promote seizure. Here, we examined pyramidal neurons lacking Na1.2 channels and found that they were intrinsically hyperexcitable, firing high-frequency bursts of action potentials (APs) despite decrements in AP size and speed. Compartmental modeling and dynamic-clamp recordings revealed that Na1.2 loss prevented potassium channels from properly repolarizing neurons between APs, increasing overall excitability by allowing neurons to reach threshold for subsequent APs more rapidly. This cell-intrinsic mechanism may, therefore, account for why SCN2A loss-of-function can paradoxically promote seizure.
编码钠通道 Na1.2 的基因 SCN2A 的功能丧失变异与自闭症谱系障碍和智力残疾密切相关。据估计,这些变异的儿童中有 20%-30%也患有癫痫,神经元活动的改变起源于新皮层,Na1.2 通道主要在兴奋性锥体神经元中表达。这是矛盾的,因为兴奋性细胞中钠通道的丧失预计会抑制新皮层的活动,而不是促进癫痫发作。在这里,我们研究了缺乏 Na1.2 通道的锥体神经元,发现它们本身就过度兴奋,尽管动作电位 (AP) 的大小和速度下降,但仍能发出高频 AP 爆发。分室建模和动态钳记录显示,Na1.2 的缺失阻止了钾通道在 AP 之间正确复极化神经元,通过使神经元更快地达到后续 AP 的阈值,从而增加整体兴奋性。因此,这种细胞内机制可能解释了为什么 SCN2A 功能丧失性变异可以反常地促进癫痫发作。