Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University, West Lafayette, IN 47907, USA; Purdue Institute for Integrative Neuroscience, Purdue University, West Lafayette, IN 47907, USA.
Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University, West Lafayette, IN 47907, USA; Purdue Institute for Integrative Neuroscience, Purdue University, West Lafayette, IN 47907, USA; Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN 47907, USA.
Cell Rep. 2021 Aug 3;36(5):109495. doi: 10.1016/j.celrep.2021.109495.
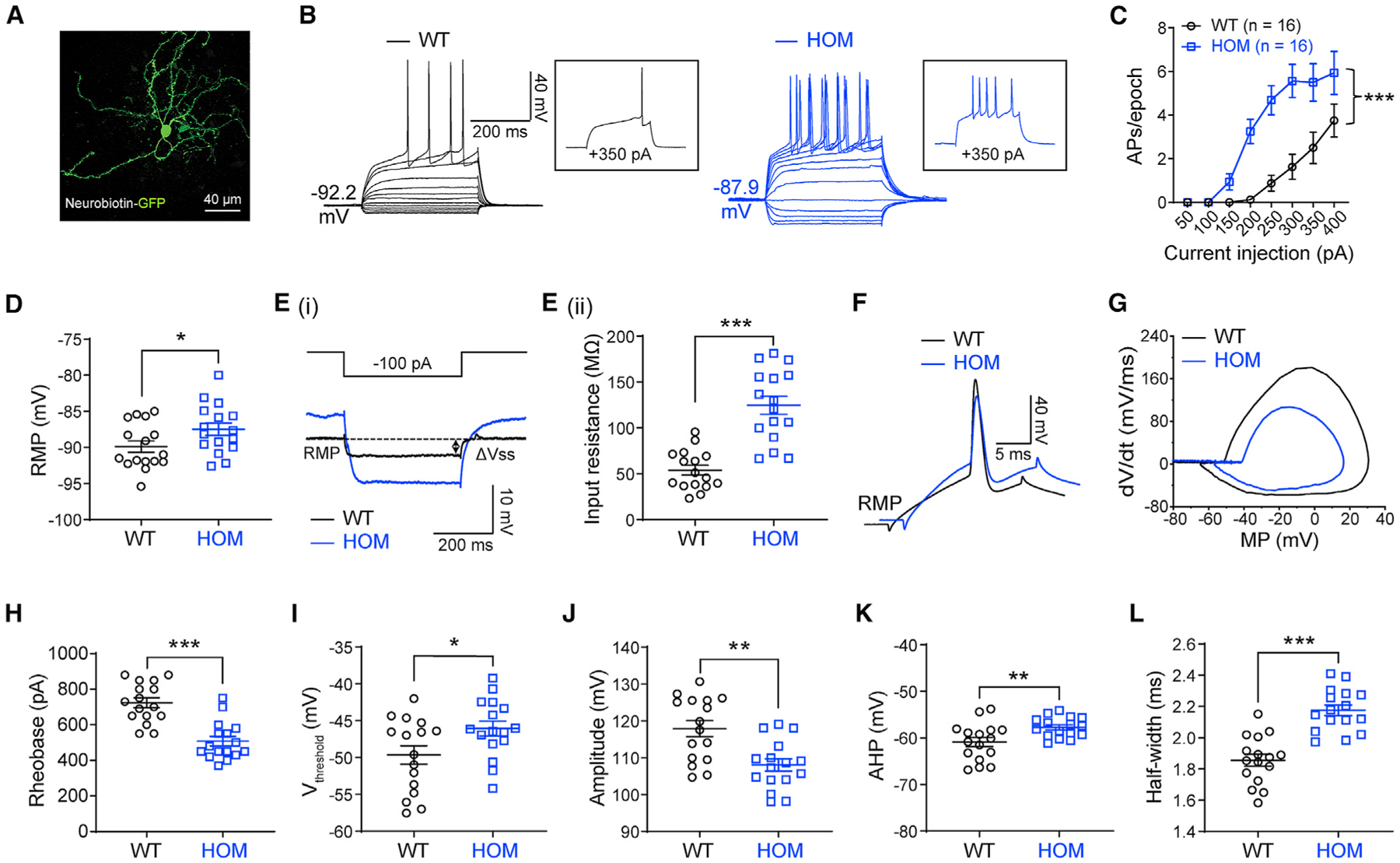
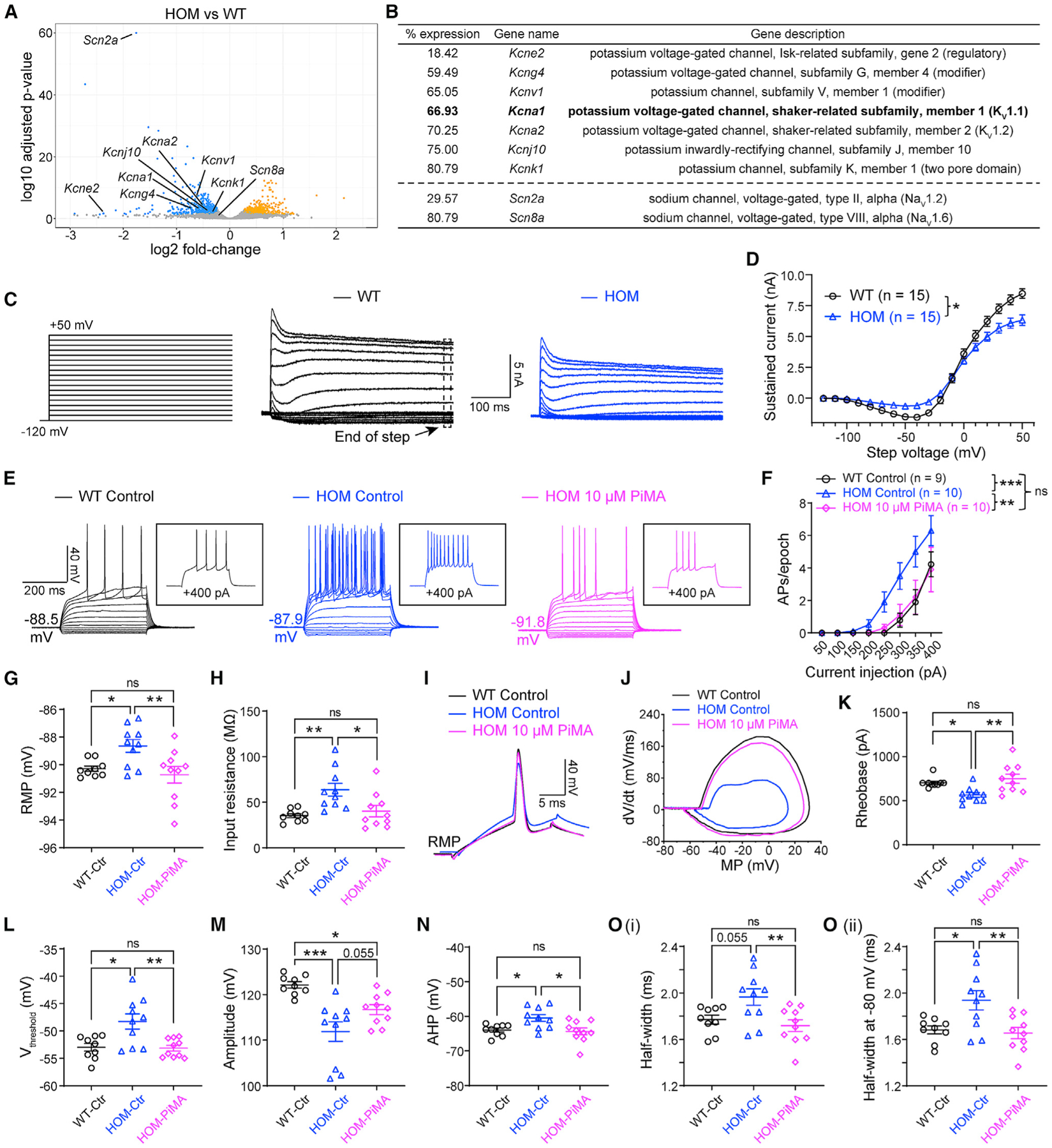
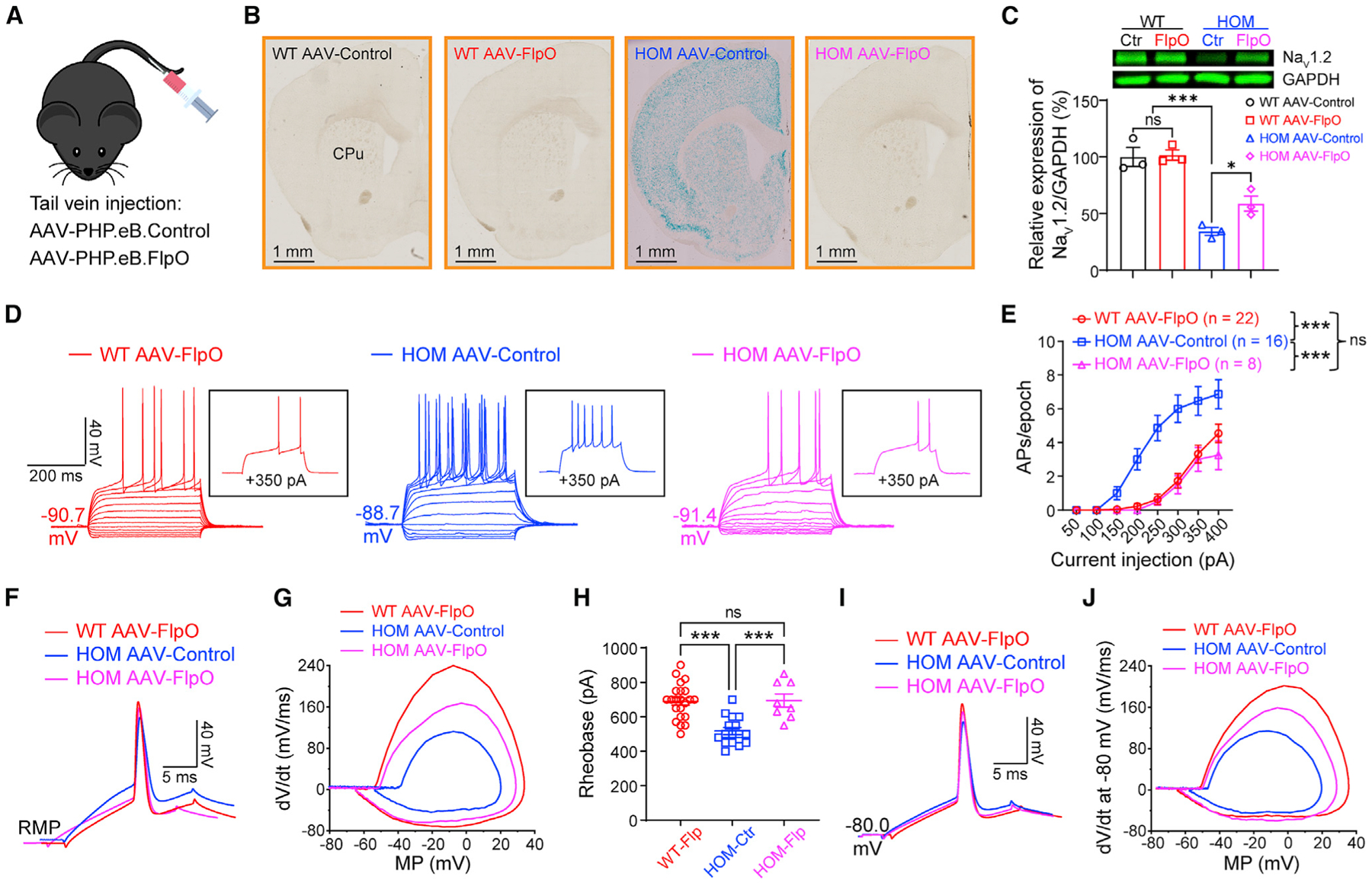
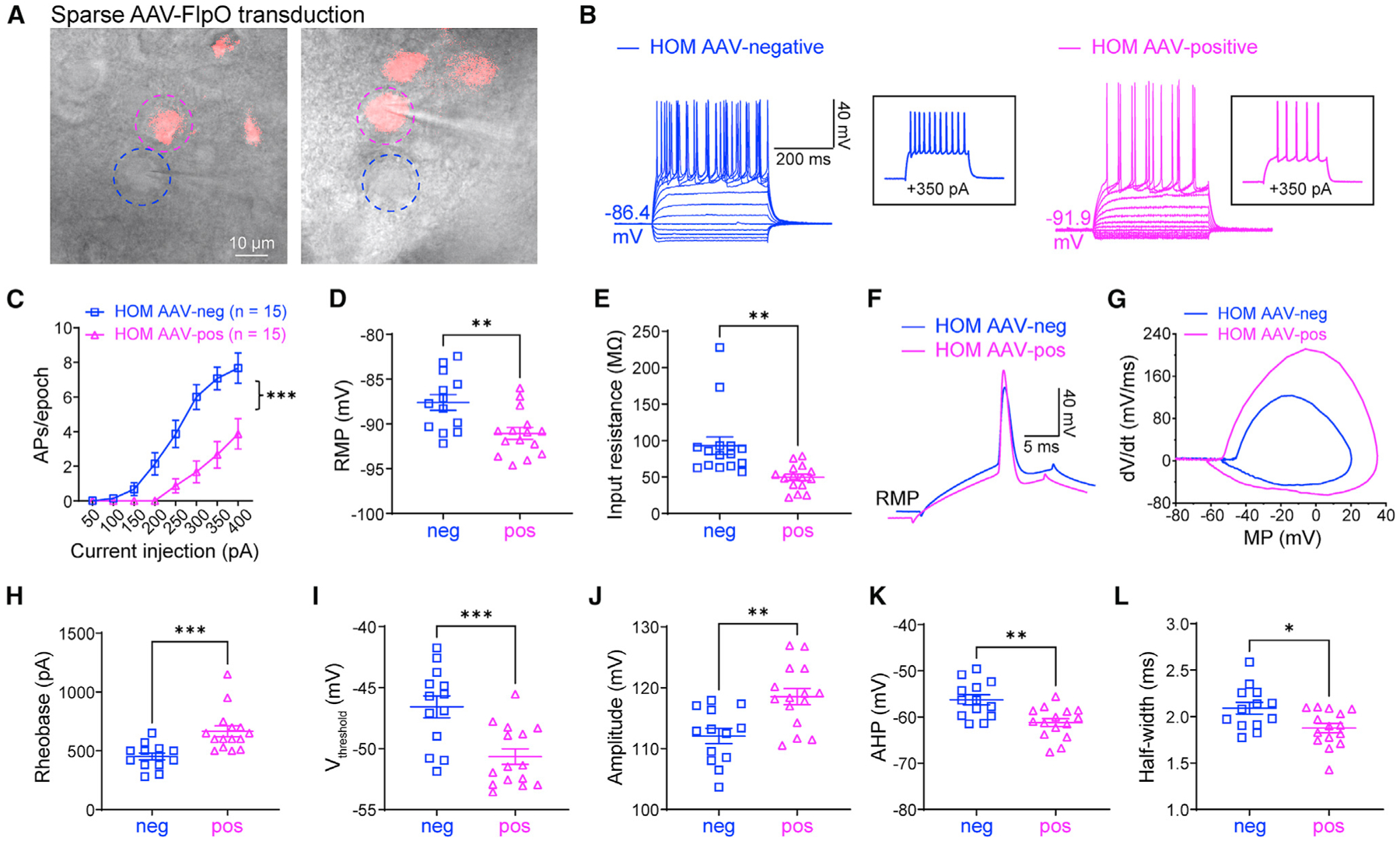
Scn2a encodes the voltage-gated sodium channel Na1.2, a main mediator of neuronal action potential firing. The current paradigm suggests that Na1.2 gain-of-function variants enhance neuronal excitability, resulting in epilepsy, whereas Na1.2 deficiency impairs neuronal excitability, contributing to autism. However, this paradigm does not explain why ∼20%-30% of individuals with Na1.2 deficiency still develop seizures. Here, we report the counterintuitive finding that severe Na1.2 deficiency results in increased neuronal excitability. Using a Na1.2-deficient mouse model, we show enhanced intrinsic excitability of principal neurons in the prefrontal cortex and striatum, brain regions known to be involved in Scn2a-related seizures. This increased excitability is autonomous and reversible by genetic restoration of Scn2a expression in adult mice. RNA sequencing reveals downregulation of multiple potassium channels, including K1.1. Correspondingly, K channel openers alleviate the hyperexcitability of Na1.2-deficient neurons. This unexpected neuronal hyperexcitability may serve as a cellular basis underlying Na1.2 deficiency-related seizures.
Scn2a 编码电压门控钠离子通道 Na1.2,Na1.2 是神经元动作电位放电的主要介导者。目前的范式表明,Na1.2 功能获得性变体增强神经元兴奋性,导致癫痫,而 Na1.2 缺乏则损害神经元兴奋性,导致自闭症。然而,这一范式并不能解释为什么大约 20%-30%的 Na1.2 缺乏患者仍然会出现癫痫发作。在这里,我们报告了一个意想不到的发现,即严重的 Na1.2 缺乏会导致神经元兴奋性增加。我们使用 Na1.2 缺乏的小鼠模型,显示出前额叶皮层和纹状体的主要神经元的内在兴奋性增强,这些脑区已知与 Scn2a 相关的癫痫发作有关。这种兴奋性增加是自主的,并且可以通过成年小鼠中 Scn2a 表达的遗传恢复来逆转。RNA 测序显示,多种钾通道包括 K1.1 的表达下调。相应地,K 通道开放剂缓解了 Na1.2 缺乏神经元的过度兴奋。这种意想不到的神经元过度兴奋可能是 Na1.2 缺乏相关癫痫发作的细胞基础。