Department of Systems and Computational Biology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY, 10461, USA.
BMC Bioinformatics. 2021 Aug 17;22(1):408. doi: 10.1186/s12859-021-04323-0.
Proteins form various complexes to carry out their versatile functions in cells. The dynamic properties of protein complex formation are mainly characterized by the association rates which measures how fast these complexes can be formed. It was experimentally observed that the association rates span an extremely wide range with over ten orders of magnitudes. Identification of association rates within this spectrum for specific protein complexes is therefore essential for us to understand their functional roles.
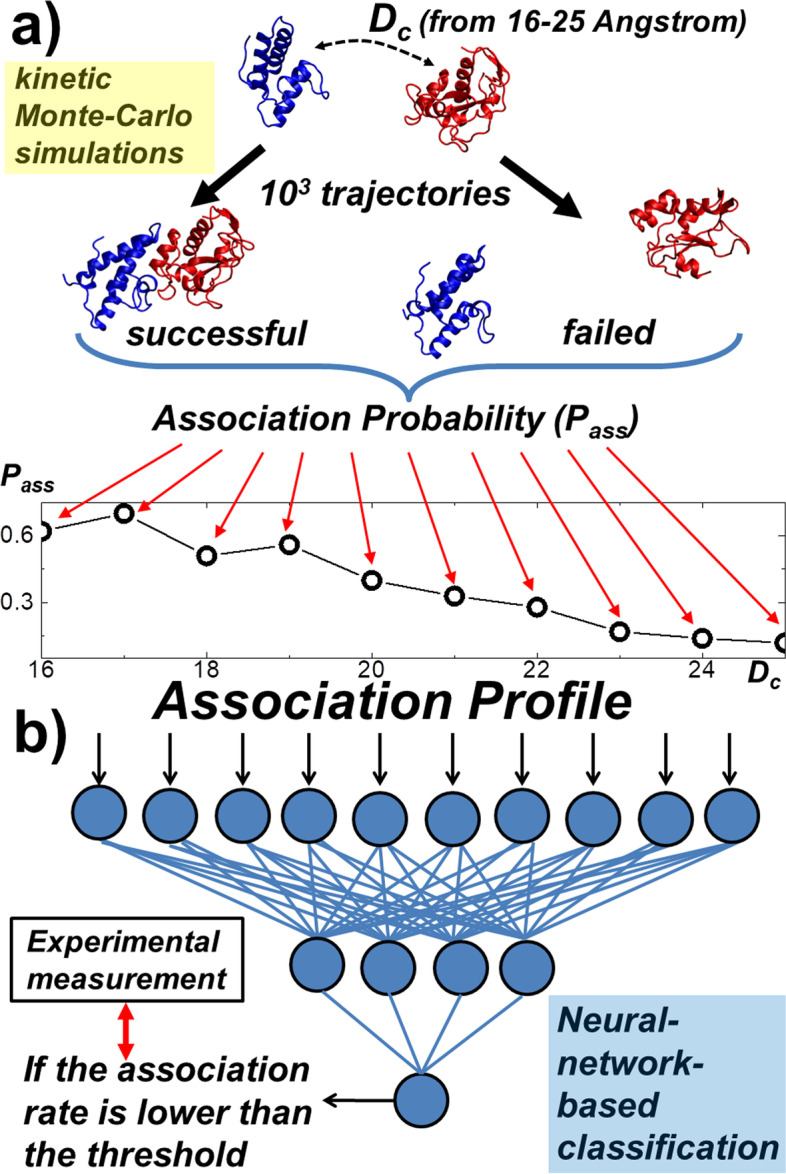
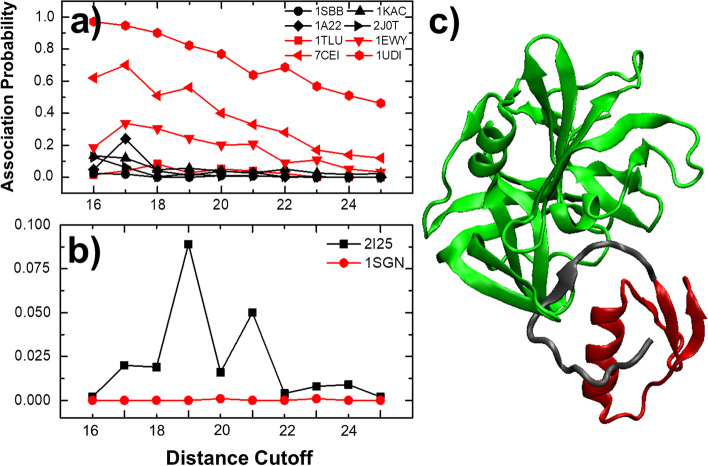
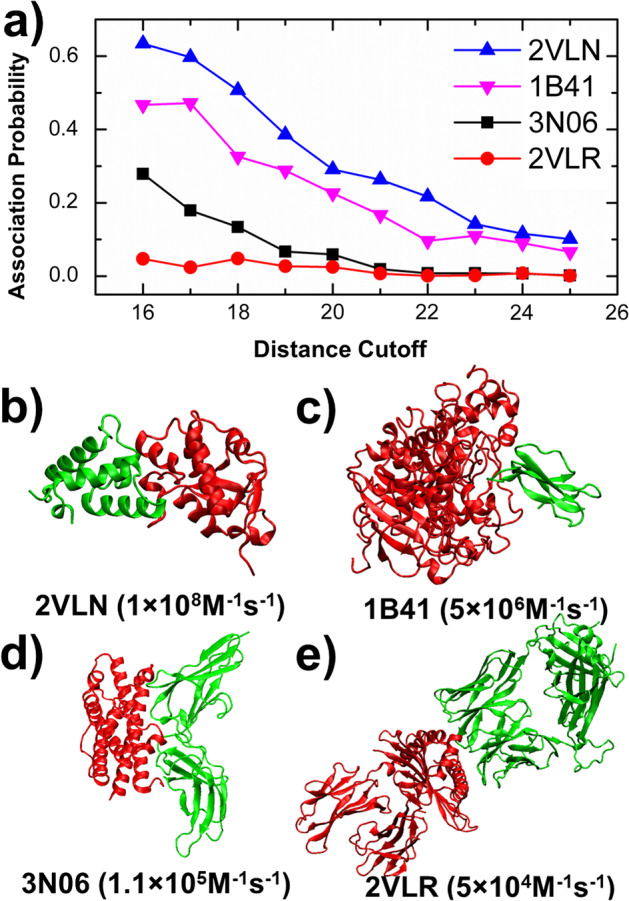
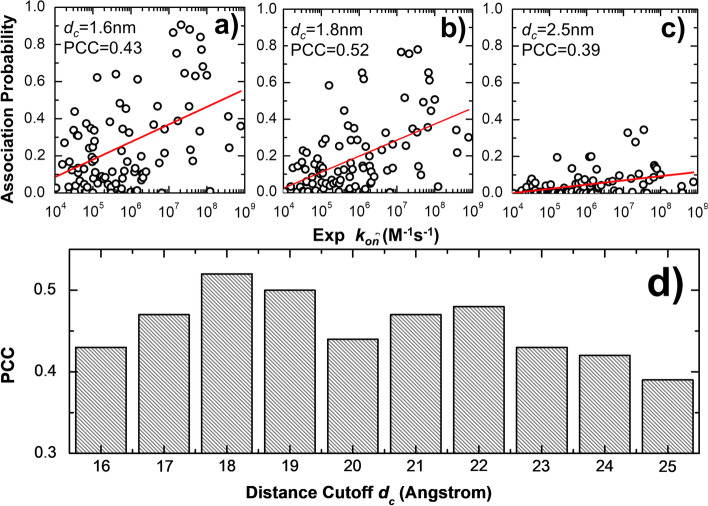
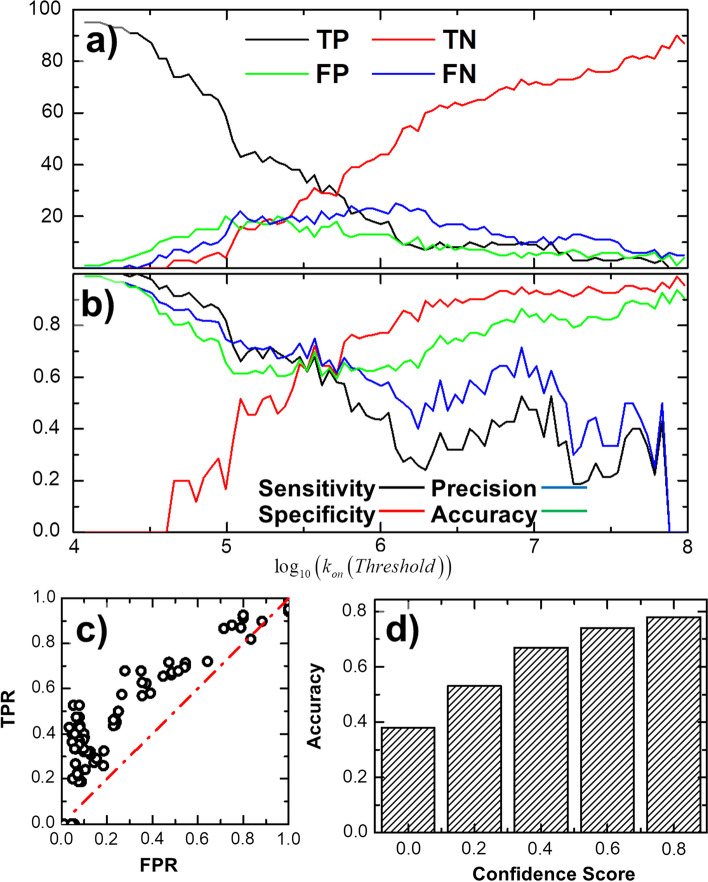
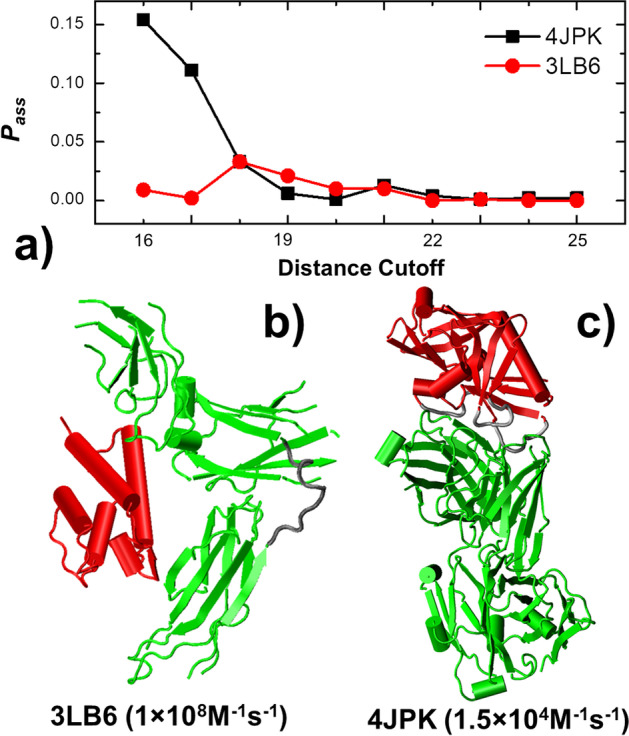
To tackle this problem, we integrate physics-based coarse-grained simulations into a neural-network-based classification model to estimate the range of association rates for protein complexes in a large-scale benchmark set. The cross-validation results show that, when an optimal threshold was selected, we can reach the best performance with specificity, precision, sensitivity and overall accuracy all higher than 70%. The quality of our cross-validation data has also been testified by further statistical analysis. Additionally, given an independent testing set, we can successfully predict the group of association rates for eight protein complexes out of ten. Finally, the analysis of failed cases suggests the future implementation of conformational dynamics into simulation can further improve model.
In summary, this study demonstrated that a new modeling framework that combines biophysical simulations with bioinformatics approaches is able to identify protein-protein interactions with low association rates from those with higher association rates. This method thereby can serve as a useful addition to a collection of existing experimental approaches that measure biomolecular recognition.
蛋白质形成各种复合物以在细胞中发挥其多种功能。蛋白质复合物形成的动态特性主要由缔合速率来表征,该速率衡量这些复合物形成的速度有多快。实验观察到,缔合速率的跨度非常大,有十个数量级以上。因此,确定特定蛋白质复合物在该范围内的缔合速率对于我们理解其功能作用至关重要。
为了解决这个问题,我们将基于物理的粗粒化模拟集成到基于神经网络的分类模型中,以估计大规模基准集中蛋白质复合物的缔合速率范围。交叉验证结果表明,当选择最佳阈值时,我们可以以特异性、精度、敏感性和整体准确性均高于 70%的最佳性能达到最佳性能。我们的交叉验证数据的质量也通过进一步的统计分析得到了验证。此外,给定一个独立的测试集,我们可以成功预测十个蛋白质复合物中的八个的缔合速率组。最后,对失败案例的分析表明,在模拟中进一步实施构象动力学可以进一步改进模型。
总之,本研究表明,将生物物理模拟与生物信息学方法相结合的新建模框架能够从具有较高缔合速率的复合物中识别具有较低缔合速率的蛋白质-蛋白质相互作用。因此,该方法可以作为现有测量生物分子识别的实验方法的有用补充。