Wang Zhongyu, Li Juan, Wang Anqi, Wang Zhaoyang, Wang Junmin, Yuan Jingjing, Wei Xin, Xing Fei, Zhang Wei, Xing Na
Department of Anesthesiology and Perioperative Medicine, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China.
Department of Human Anatomy, Basic Medical College of Zhengzhou University, Zhengzhou, China.
Front Cell Dev Biol. 2021 Aug 4;9:658720. doi: 10.3389/fcell.2021.658720. eCollection 2021.
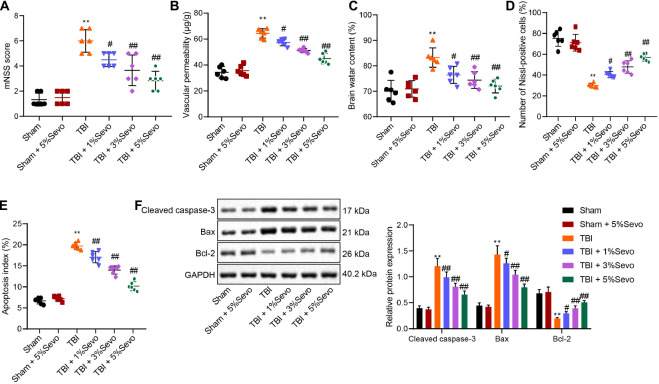
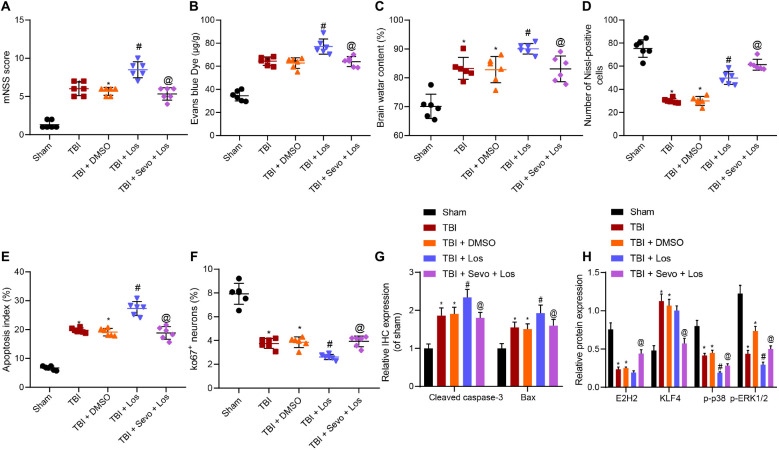
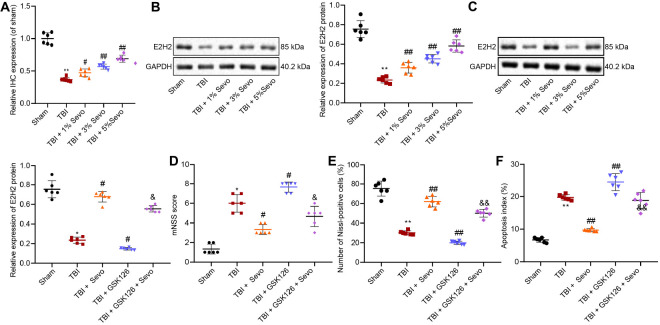
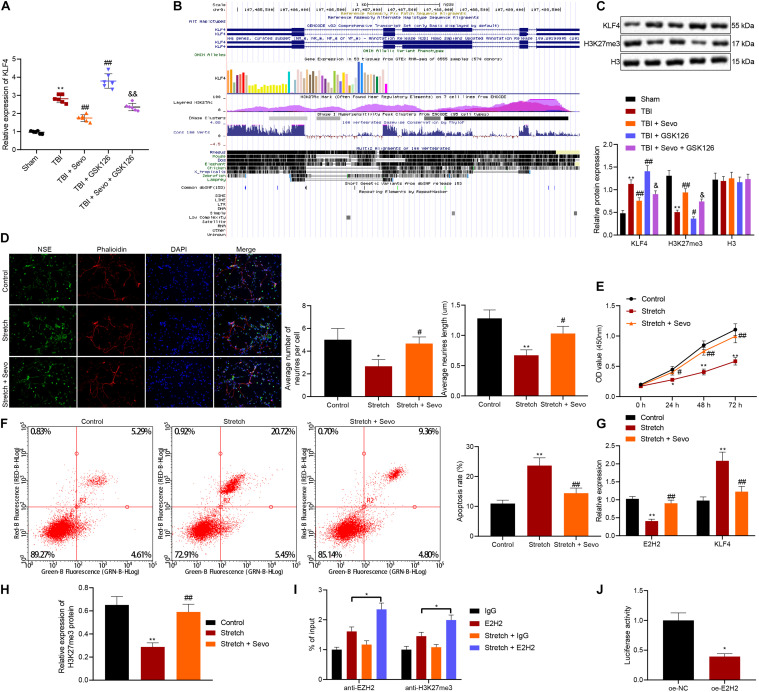
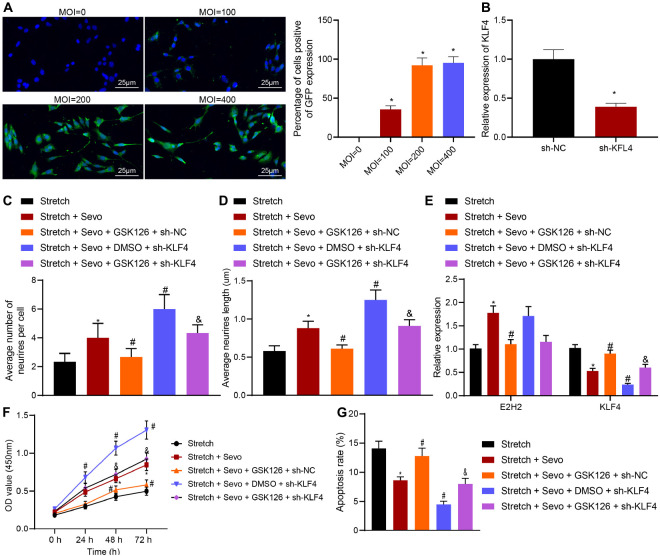
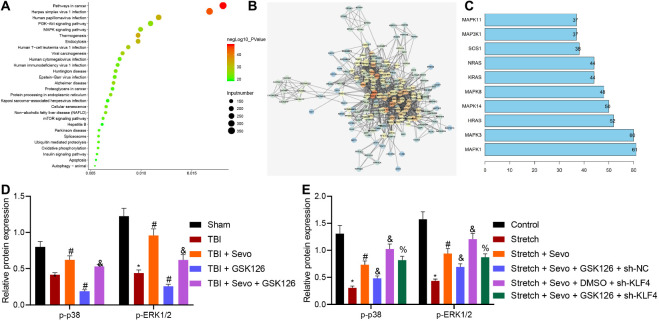
Traumatic brain injury (TBI) is characterized by physical damage to the brain tissues, ensuing transitory or permanent neurological dysfunction featured with neuronal loss and subsequent brain damage. Sevoflurane, a widely used halogenated anesthetic in clinical settings, has been reported to alleviate neuron apoptosis in TBI. Nevertheless, the underlying mechanism behind this alleviation remains unknown, and thus was the focus of the current study. First, Feeney models were established to induce TBI in rats. Subsequently, evaluation of the modified neurological severity scores, measurement of brain water content, Nissl staining, and TUNEL assay were employed to investigate the neuroprotective effects of sevoflurane. Immunofluorescence and Western blot analysis were further applied to detect the expression patterns of apoptosis-related proteins as well as the activation of the p38-mitogen-activated protein kinase (MAPK) signaling pathway within the lesioned cortex. Additionally, a stretch injury model comprising cultured neurons was established, followed by neuron-specific enolase staining and Sholl analysis. Mechanistic analyses were performed using dual-luciferase reporter gene and chromatin immunoprecipitation assays. The results demonstrated sevoflurane treatment brought about a decrease blood-brain barrier (BBB) permeability, brain water content, brain injury and neuron apoptosis, to improve neurological function. The neuroprotective action of sevoflurane could be attenuated by inactivation of the p38-MAPK signaling pathway. Mechanistically, sevoflurane exerted an inhibitory effect on neuron apoptosis by up-regulating enhancer of zeste homolog 2 (EZH2), which targeted Krüppel-like factor 4 (KLF4) and inhibited KLF4 transcription. Collectively, our findings indicate that sevoflurane suppresses neuron apoptosis induced by TBI through activation of the p38-MAPK signaling pathway the EZH2/KLF4 axis, providing a novel mechanistic explanation for neuroprotection of sevoflurane in TBI.
创伤性脑损伤(TBI)的特征是脑组织受到物理损伤,继而出现以神经元丢失和随后的脑损伤为特征的短暂或永久性神经功能障碍。七氟醚是临床环境中广泛使用的卤化麻醉剂,据报道可减轻TBI中的神经元凋亡。然而,这种减轻背后的潜在机制仍然未知,因此是本研究的重点。首先,建立Feeney模型以诱导大鼠TBI。随后,采用改良神经功能缺损评分评估、脑含水量测量、尼氏染色和TUNEL检测来研究七氟醚的神经保护作用。进一步应用免疫荧光和蛋白质印迹分析来检测损伤皮层中凋亡相关蛋白的表达模式以及p38丝裂原活化蛋白激酶(MAPK)信号通路的激活。此外,建立了包含培养神经元的拉伸损伤模型,随后进行神经元特异性烯醇化酶染色和Sholl分析。使用双荧光素酶报告基因和染色质免疫沉淀试验进行机制分析。结果表明,七氟醚治疗可降低血脑屏障(BBB)通透性、脑含水量、脑损伤和神经元凋亡,改善神经功能。七氟醚的神经保护作用可通过p38-MAPK信号通路失活而减弱。机制上,七氟醚通过上调zeste同源物2(EZH2)增强子发挥对神经元凋亡的抑制作用,EZH2靶向Krüppel样因子4(KLF4)并抑制KLF4转录。总的来说,我们的研究结果表明,七氟醚通过激活p38-MAPK信号通路-EZH2/KLF4轴抑制TBI诱导的神经元凋亡,为七氟醚在TBI中的神经保护作用提供了新的机制解释。