Faculty of Pharmacy, University of Ljubljana, 1000 Ljubljana, Slovenia.
Theory Department, National Institute of Chemistry, 1000 Ljubljana, Slovenia.
Int J Mol Sci. 2021 Aug 20;22(16):8999. doi: 10.3390/ijms22168999.
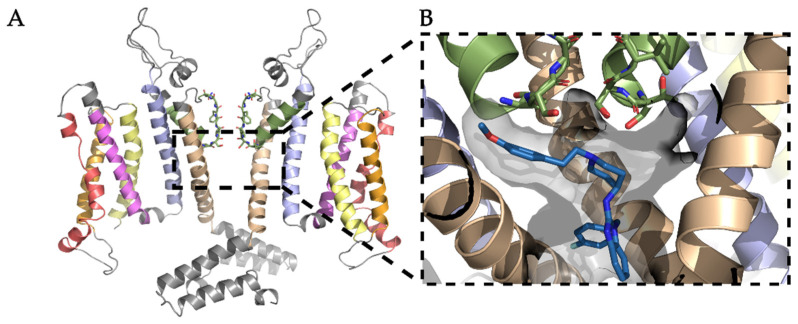
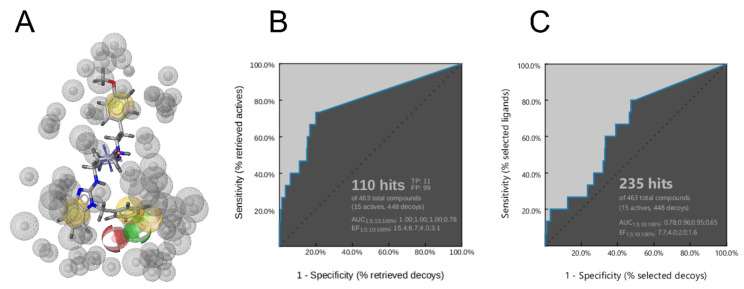
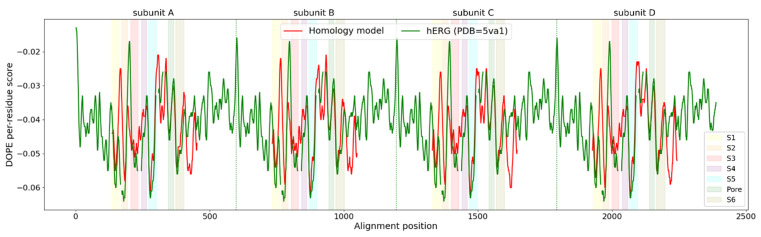
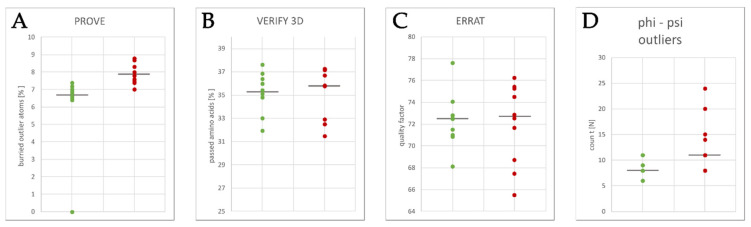
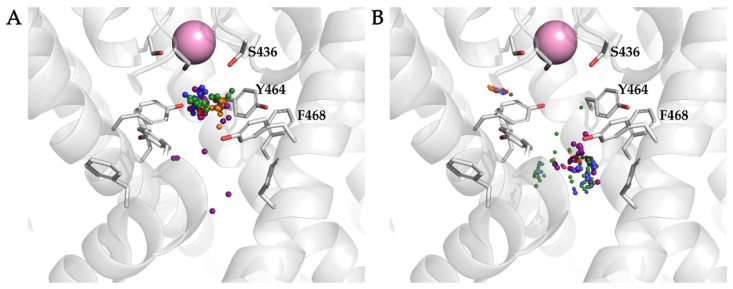
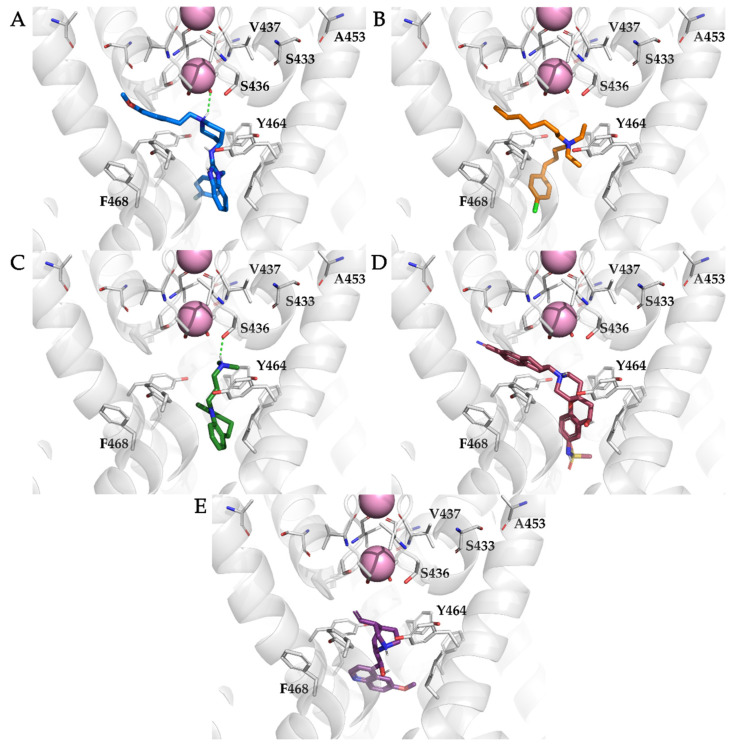
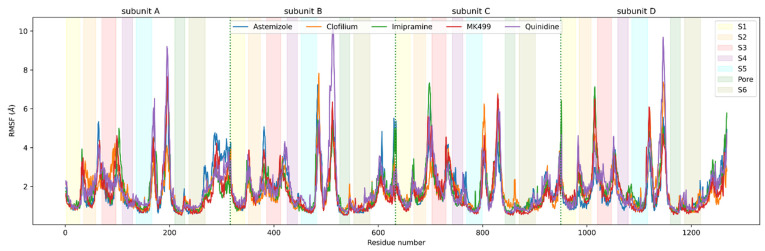
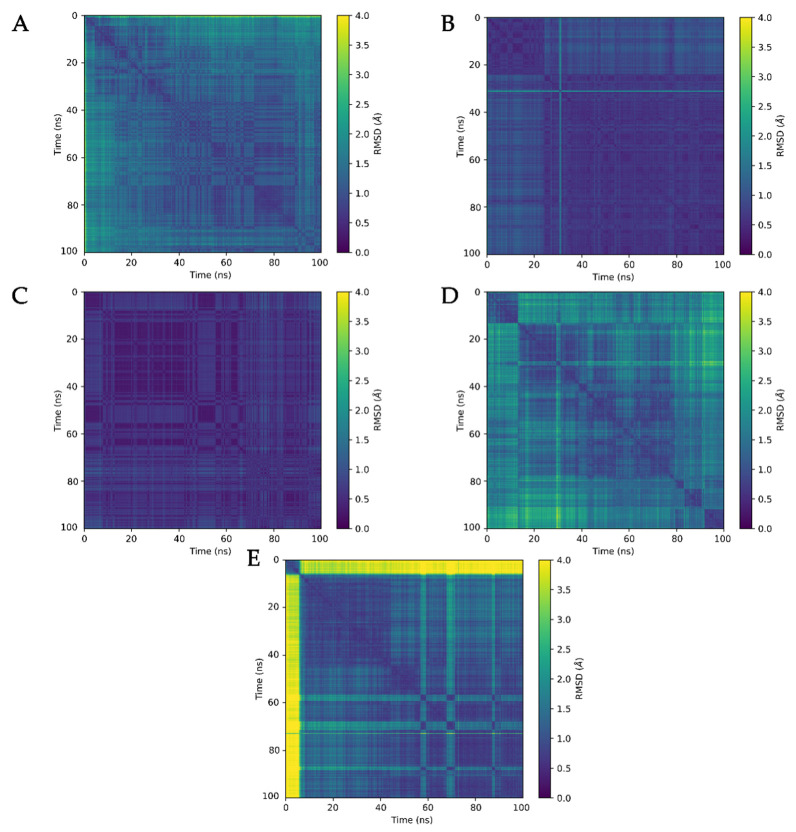
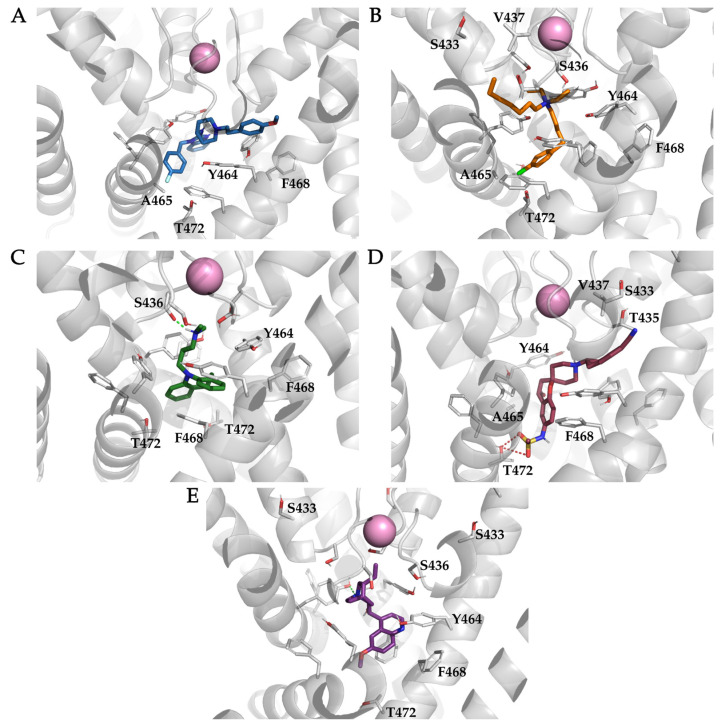
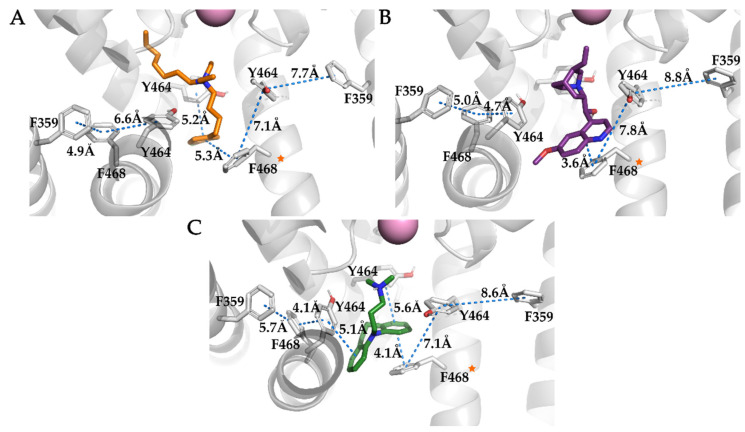
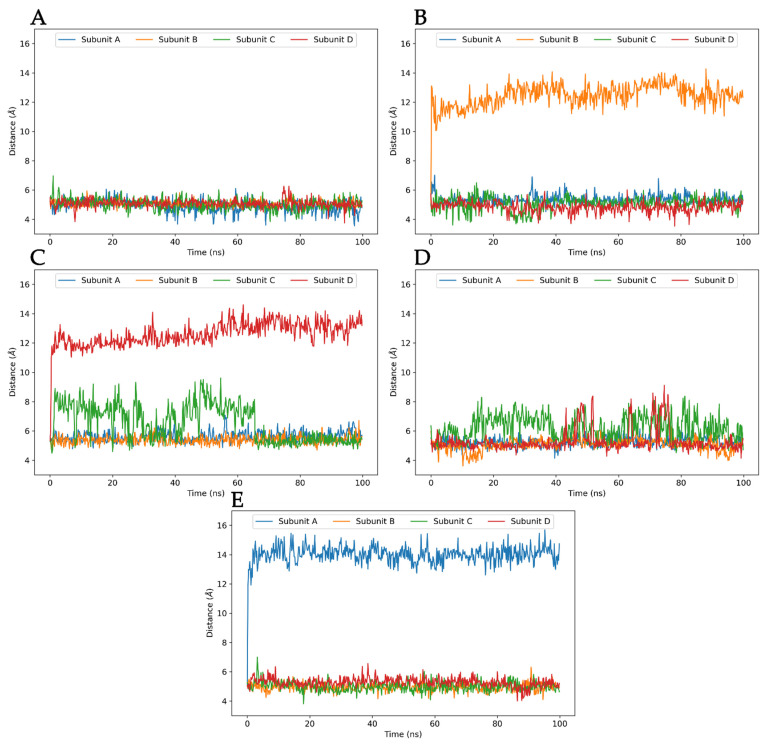
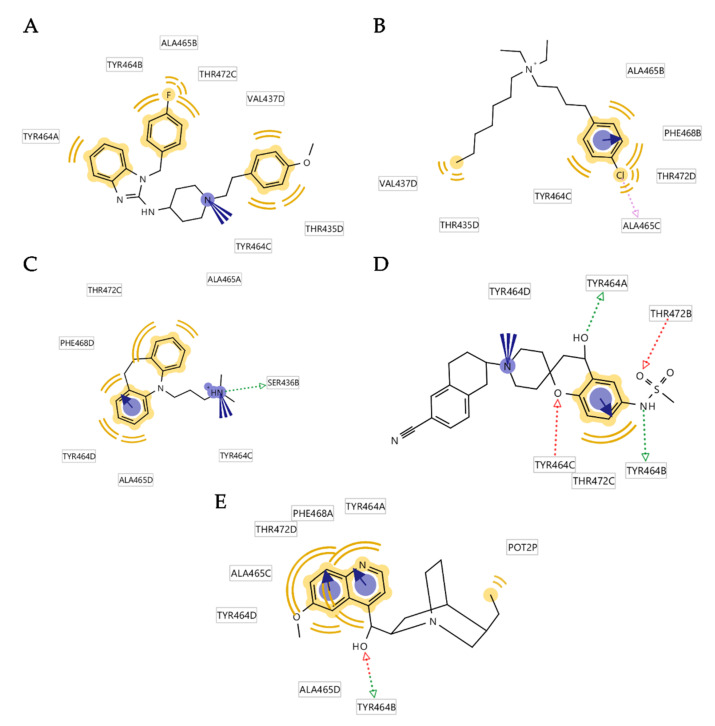
The K10.1 voltage-gated potassium channel is highly expressed in 70% of tumors, and thus represents a promising target for anticancer drug discovery. However, only a few ligands are known to inhibit K10.1, and almost all also inhibit the very similar cardiac hERG channel, which can lead to undesirable side-effects. In the absence of the structure of the K10.1-inhibitor complex, there remains the need for new strategies to identify selective K10.1 inhibitors and to understand the binding modes of the known K10.1 inhibitors. To investigate these binding modes in the central cavity of K10.1, a unique approach was used that allows derivation and analysis of ligand-protein interactions from molecular dynamics trajectories through pharmacophore modeling. The final molecular dynamics-derived structure-based pharmacophore model for the simulated K10.1-ligand complexes describes the necessary pharmacophore features for K10.1 inhibition and is highly similar to the previously reported ligand-based hERG pharmacophore model used to explain the nonselectivity of K10.1 pore blockers. Moreover, analysis of the molecular dynamics trajectories revealed disruption of the π-π network of aromatic residues F359, Y464, and F468 of K10.1, which has been reported to be important for binding of various ligands for both K10.1 and hERG channels. These data indicate that targeting the K10.1 channel pore is also likely to result in undesired hERG inhibition, and other potential binding sites should be explored to develop true K10.1-selective inhibitors as new anticancer agents.
K10.1 电压门控钾通道在 70%的肿瘤中高度表达,因此代表了抗癌药物发现的有前途的靶标。然而,已知只有少数配体能够抑制 K10.1,并且几乎所有配体也抑制非常相似的心脏 hERG 通道,这可能导致不良的副作用。在缺乏 K10.1-抑制剂复合物的结构的情况下,仍然需要新的策略来识别选择性 K10.1 抑制剂并了解已知 K10.1 抑制剂的结合模式。为了研究 K10.1 中央腔中的这些结合模式,采用了一种独特的方法,该方法允许通过药效团建模从分子动力学轨迹中推导出和分析配体-蛋白相互作用。最终的基于分子动力学的结构药效团模型用于模拟 K10.1-配体复合物,描述了 K10.1 抑制所必需的药效团特征,并且与先前报道的用于解释 K10.1 孔阻滞剂非选择性的基于配体的 hERG 药效团模型高度相似。此外,对分子动力学轨迹的分析揭示了 K10.1 中芳香残基 F359、Y464 和 F468 的 π-π 网络的破坏,据报道该网络对于各种配体与 K10.1 和 hERG 通道的结合都很重要。这些数据表明,靶向 K10.1 通道孔也可能导致不期望的 hERG 抑制,并且应该探索其他潜在的结合位点,以开发真正的 K10.1 选择性抑制剂作为新的抗癌药物。