Department of Medical Genetics, St. Olavs Hospital, Trondheim, Norway.
Department of Clinical and Molecular Medicine, Faculty of Medicine and Health Sciences, NTNU - Norwegian University of Science and Technology, Trondheim, Norway.
BMC Med Genomics. 2021 Aug 31;14(1):214. doi: 10.1186/s12920-021-01059-x.
Detection of copy number variation (CNV) in genes associated with disease is important in genetic diagnostics, and next generation sequencing (NGS) technology provides data that can be used for CNV detection. However, CNV detection based on NGS data is in general not often used in diagnostic labs as the data analysis is challenging, especially with data from targeted gene panels. Wet lab methods like MLPA (MRC Holland) are widely used, but are expensive, time consuming and have gene-specific limitations. Our aim has been to develop a bioinformatic tool for CNV detection from NGS data in medical genetic diagnostic samples.
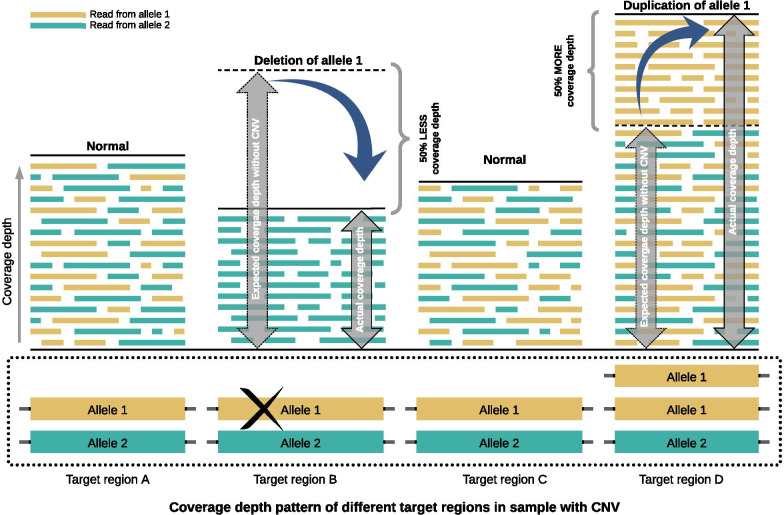
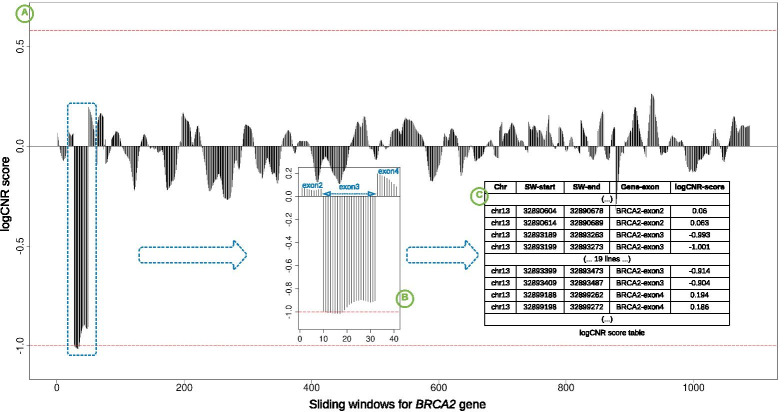
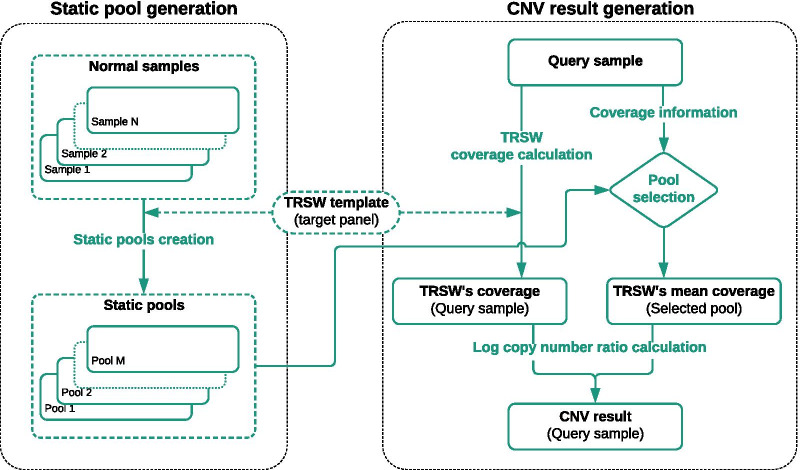
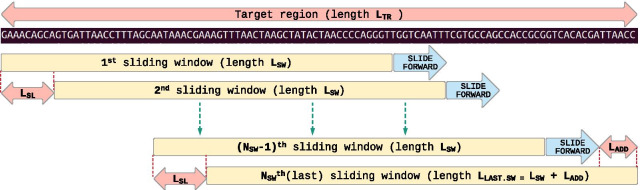
Our computational pipeline for detection of CNVs in NGS data from targeted gene panels utilizes coverage depth of the captured regions and calculates a copy number ratio score for each region. This is computed by comparing the mean coverage of the sample with the mean coverage of the same region in other samples, defined as a pool. The pipeline selects pools for comparison dynamically from previously sequenced samples, using the pool with an average coverage depth that is nearest to the one of the samples. A sliding window-based approach is used to analyze each region, where length of sliding window and sliding distance can be chosen dynamically to increase or decrease the resolution. This helps in detecting CNVs in small or partial exons. With this pipeline we have correctly identified the CNVs in 36 positive control samples, with sensitivity of 100% and specificity of 91%. We have detected whole gene level deletion/duplication, single/multi exonic level deletion/duplication, partial exonic deletion and mosaic deletion. Since its implementation in mid-2018 it has proven its diagnostic value with more than 45 CNV findings in routine tests.
With this pipeline as part of our diagnostic practices it is now possible to detect partial, single or multi-exonic, and intragenic CNVs in all genes in our target panel. This has helped our diagnostic lab to expand the portfolio of genes where we offer CNV detection, which previously was limited by the availability of MLPA kits.
在遗传诊断中,检测与疾病相关的基因拷贝数变异(CNV)很重要,下一代测序(NGS)技术提供了可用于 CNV 检测的数据。然而,由于数据分析具有挑战性,特别是针对靶向基因panel 的 NGS 数据,基于 NGS 数据的 CNV 检测通常未在诊断实验室中广泛应用。Wet lab 方法如 MLPA(MRC Holland)被广泛应用,但昂贵、耗时且具有基因特异性限制。我们的目标是开发一种用于从医学遗传诊断样本中的 NGS 数据中检测 CNV 的生物信息学工具。
我们用于从靶向基因panel 的 NGS 数据中检测 CNV 的计算管道利用了捕获区域的覆盖深度,并为每个区域计算了拷贝数比得分。这是通过比较样本的平均覆盖度与其他样本中同一区域的平均覆盖度来计算的,这些样本定义为一个pool。该管道使用之前测序的样本中动态选择用于比较的 pool,使用与样本平均覆盖度最接近的 pool。使用基于滑动窗口的方法来分析每个区域,其中滑动窗口的长度和滑动距离可以动态选择以增加或减少分辨率。这有助于检测小的或部分外显子中的 CNV。使用该管道,我们在 36 个阳性对照样本中正确识别出了 CNV,敏感性为 100%,特异性为 91%。我们已经检测到了全基因水平的缺失/重复、单/多外显子水平的缺失/重复、部分外显子缺失和镶嵌缺失。自 2018 年年中实施以来,它已经通过在常规测试中发现了 45 多个 CNV 发现证明了其诊断价值。
通过将该管道纳入我们的诊断实践,现在可以在我们的目标panel 中的所有基因中检测部分、单或多外显子和基因内的 CNV。这帮助我们的诊断实验室扩大了我们提供 CNV 检测的基因组合,而以前这受到 MLPA 试剂盒可用性的限制。