Bioinformatics Interdepartmental Program, University of California, Los Angeles, Los Angeles, CA, USA.
Computational and Systems Biology, University of California, Los Angeles, Los Angeles, CA, USA.
Mol Syst Biol. 2021 Sep;17(9):e10243. doi: 10.15252/msb.202110243.
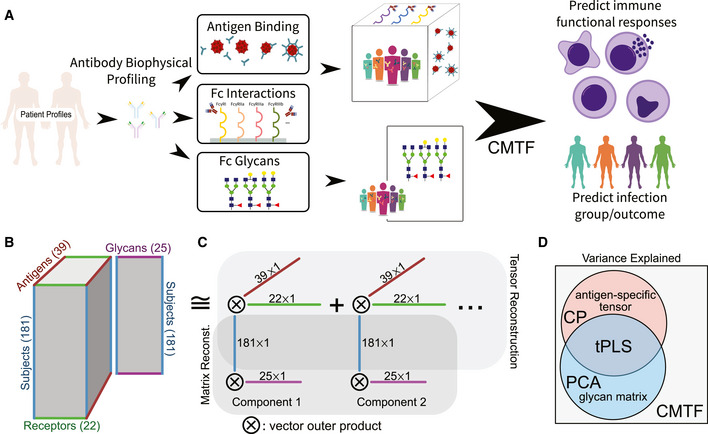
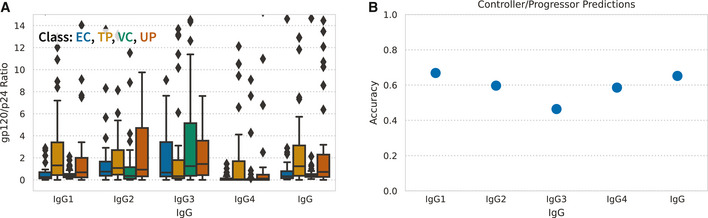
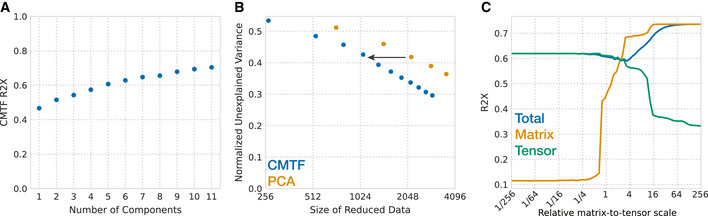
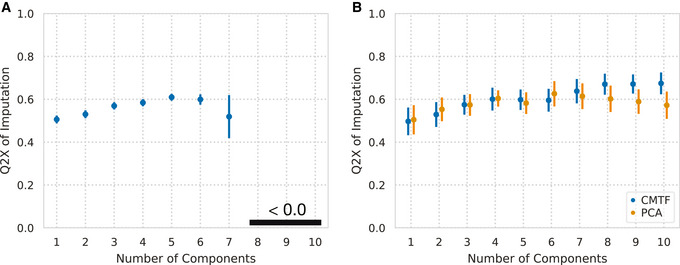
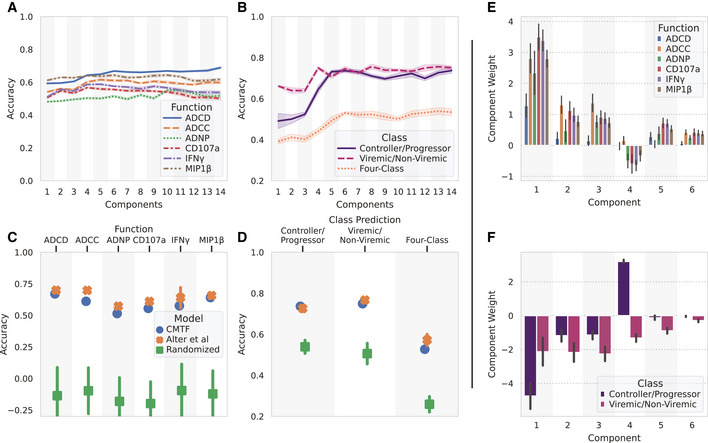
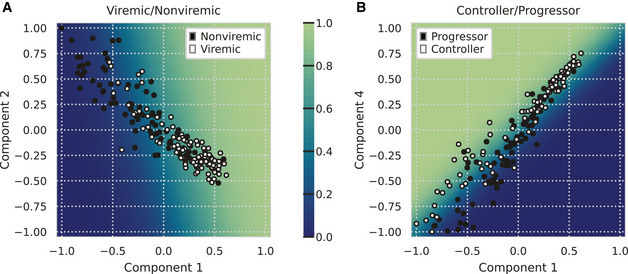
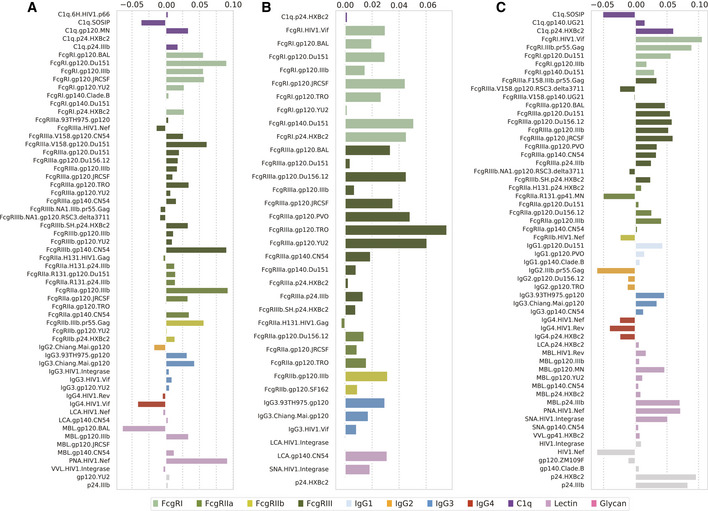
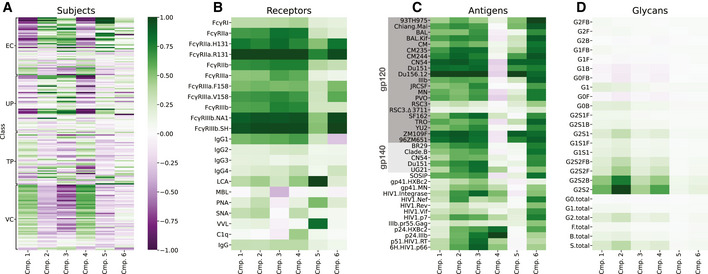
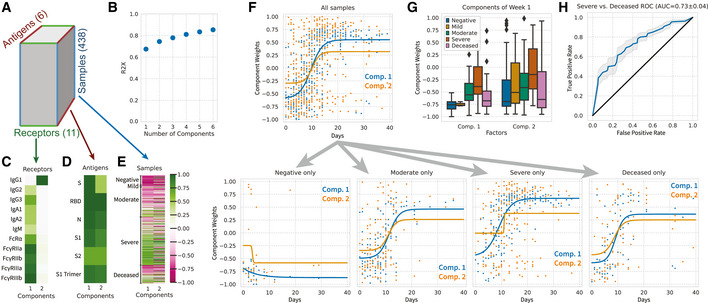
Systems serology provides a broad view of humoral immunity by profiling both the antigen-binding and Fc properties of antibodies. These studies contain structured biophysical profiling across disease-relevant antigen targets, alongside additional measurements made for single antigens or in an antigen-generic manner. Identifying patterns in these measurements helps guide vaccine and therapeutic antibody development, improve our understanding of diseases, and discover conserved regulatory mechanisms. Here, we report that coupled matrix-tensor factorization (CMTF) can reduce these data into consistent patterns by recognizing the intrinsic structure of these data. We use measurements from two previous studies of HIV- and SARS-CoV-2-infected subjects as examples. CMTF outperforms standard methods like principal components analysis in the extent of data reduction while maintaining equivalent prediction of immune functional responses and disease status. Under CMTF, model interpretation improves through effective data reduction, separation of the Fc and antigen-binding effects, and recognition of consistent patterns across individual measurements. Data reduction also helps make prediction models more replicable. Therefore, we propose that CMTF is an effective general strategy for data exploration in systems serology.
系统血清学通过分析抗体的抗原结合和 Fc 特性,提供了对体液免疫的广泛了解。这些研究包含了针对相关抗原靶标的结构生物物理分析,以及针对单个抗原或抗原通用方式进行的其他测量。识别这些测量中的模式有助于指导疫苗和治疗性抗体的开发,增进我们对疾病的理解,并发现保守的调节机制。在这里,我们报告说,通过识别这些数据的固有结构,耦合矩阵张量分解(CMTF)可以将这些数据简化为一致的模式。我们使用来自之前两项关于 HIV 和 SARS-CoV-2 感染患者的研究的测量结果作为示例。CMTF 在数据减少的程度上优于主成分分析等标准方法,同时保持对免疫功能反应和疾病状态的等效预测。在 CMTF 下,通过有效的数据减少、Fc 和抗原结合效应的分离以及跨单个测量的一致模式的识别,模型解释得到了改善。数据减少还有助于使预测模型更具可复制性。因此,我们建议 CMTF 是系统血清学中数据探索的有效通用策略。