Cancer Genetics and Comparative Genomics Branch, National Human Genome Research Institute, National Institutes of Health, 50 South Drive, Building 50, Room 5351, Bethesda, MD, 20892 , USA.
State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, 650223, China.
Mamm Genome. 2022 Mar;33(1):213-229. doi: 10.1007/s00335-021-09914-z. Epub 2021 Sep 8.
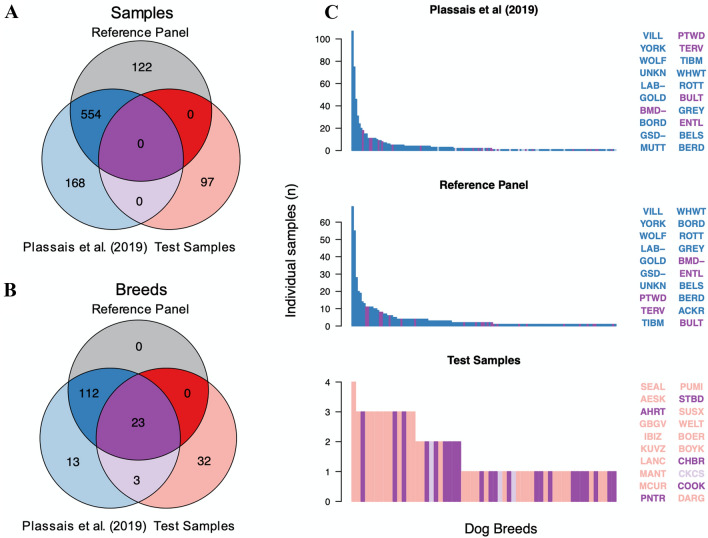
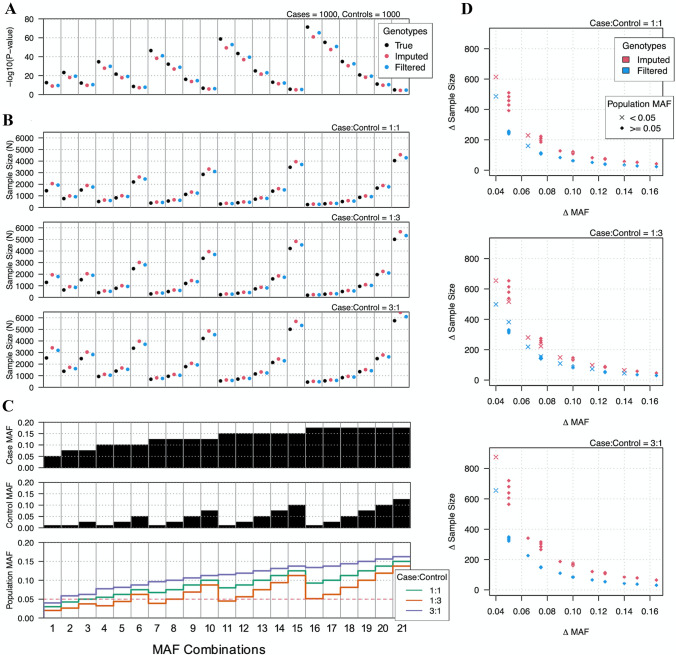
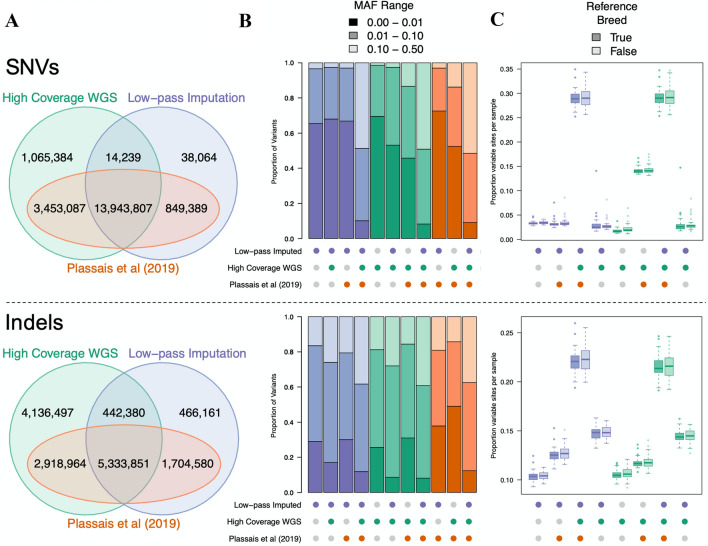
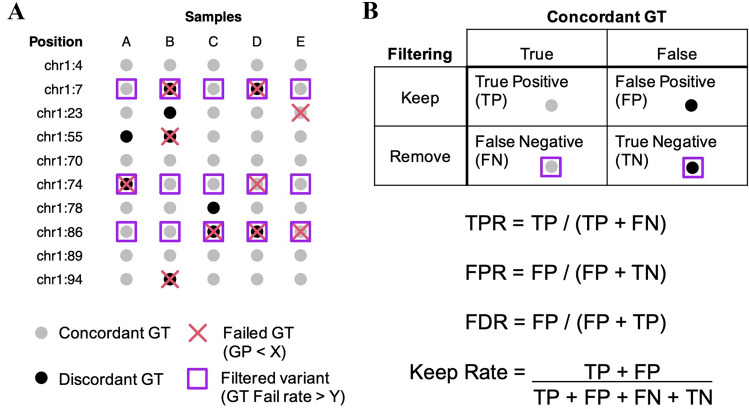
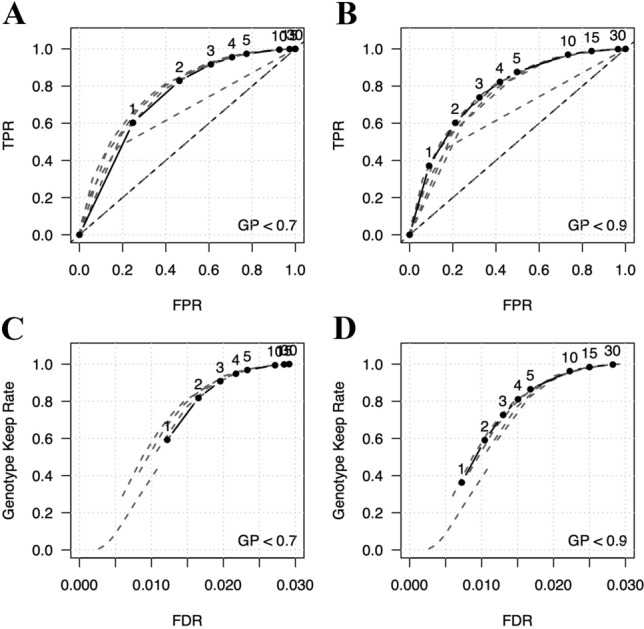
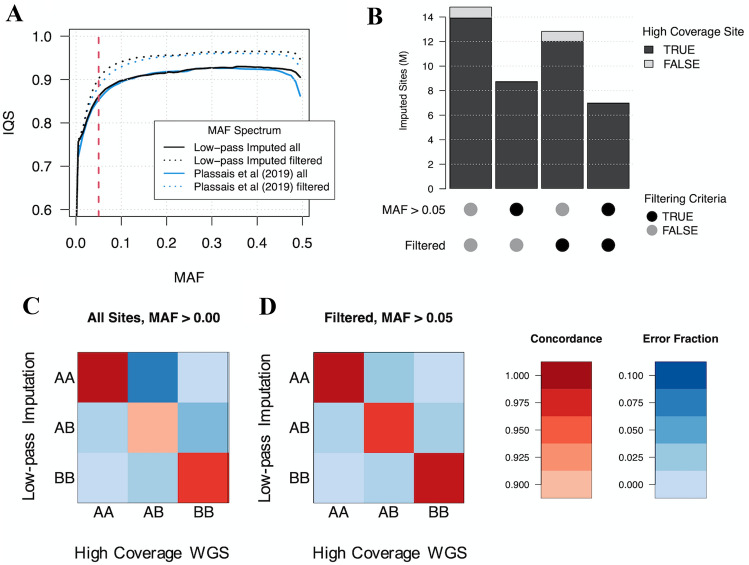
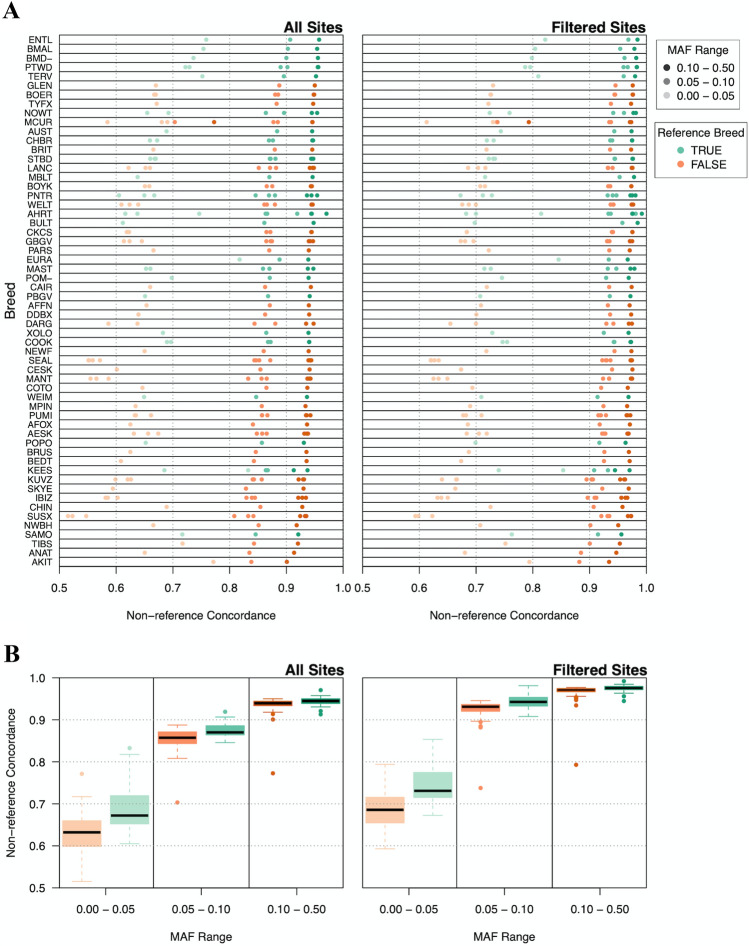
Although DNA array-based approaches for genome-wide association studies (GWAS) permit the collection of thousands of low-cost genotypes, it is often at the expense of resolution and completeness, as SNP chip technologies are ultimately limited by SNPs chosen during array development. An alternative low-cost approach is low-pass whole genome sequencing (WGS) followed by imputation. Rather than relying on high levels of genotype confidence at a set of select loci, low-pass WGS and imputation rely on the combined information from millions of randomly sampled low-confidence genotypes. To investigate low-pass WGS and imputation in the dog, we assessed accuracy and performance by downsampling 97 high-coverage (> 15×) WGS datasets from 51 different breeds to approximately 1× coverage, simulating low-pass WGS. Using a reference panel of 676 dogs from 91 breeds, genotypes were imputed from the downsampled data and compared to a truth set of genotypes generated from high-coverage WGS. Using our truth set, we optimized a variant quality filtering strategy that retained approximately 80% of 14 M imputed sites and lowered the imputation error rate from 3.0% to 1.5%. Seven million sites remained with a MAF > 5% and an average imputation quality score of 0.95. Finally, we simulated the impact of imputation errors on outcomes for case-control GWAS, where small effect sizes were most impacted and medium-to-large effect sizes were minorly impacted. These analyses provide best practice guidelines for study design and data post-processing of low-pass WGS-imputed genotypes in dogs.
尽管基于 DNA 芯片的全基因组关联研究 (GWAS) 方法允许收集数千个低成本的基因型,但这往往是以分辨率和完整性为代价的,因为 SNP 芯片技术最终受到在芯片开发过程中选择的 SNP 的限制。一种替代的低成本方法是低深度全基因组测序 (WGS) 后进行 imputation。低深度 WGS 和 imputation 不是依赖于一组精选位点的高基因型置信度,而是依赖于从数百万个随机采样的低置信度基因型中获得的综合信息。为了在犬中研究低深度 WGS 和 imputation,我们通过从 51 个不同品种中评估 97 个高覆盖率 (>15×) WGS 数据集的子集到大约 1×的覆盖率,模拟低深度 WGS,来评估准确性和性能。使用来自 91 个品种的 676 只犬的参考面板,从下采样的数据中 impute 基因型,并将其与从高覆盖率 WGS 生成的真实基因型集进行比较。使用我们的真实集,我们优化了一种变体质量过滤策略,保留了大约 80%的 1400 万个 imputed 位点,并将 imputation 错误率从 3.0%降低到 1.5%。仍有 700 万个位点具有 MAF>5%和平均 imputation 质量评分 0.95。最后,我们模拟了 imputation 错误对病例对照 GWAS 结果的影响,其中小效应大小受到的影响最大,中到大效应大小受到的影响较小。这些分析为犬的低深度 WGS-imputed 基因型的研究设计和数据后处理提供了最佳实践指南。