Laboratory of Biochemical Neuroendocrinology, Montreal Clinical Research Institute (IRCM), Affiliated to the University of Montreal, Montreal, Quebec, Canada.
Division of Nephrology, Department of Medicine, McMaster University, St. Joseph's Healthcare Hamilton, Hamilton, Ontario, Canada.
J Biol Chem. 2021 Oct;297(4):101177. doi: 10.1016/j.jbc.2021.101177. Epub 2021 Sep 8.
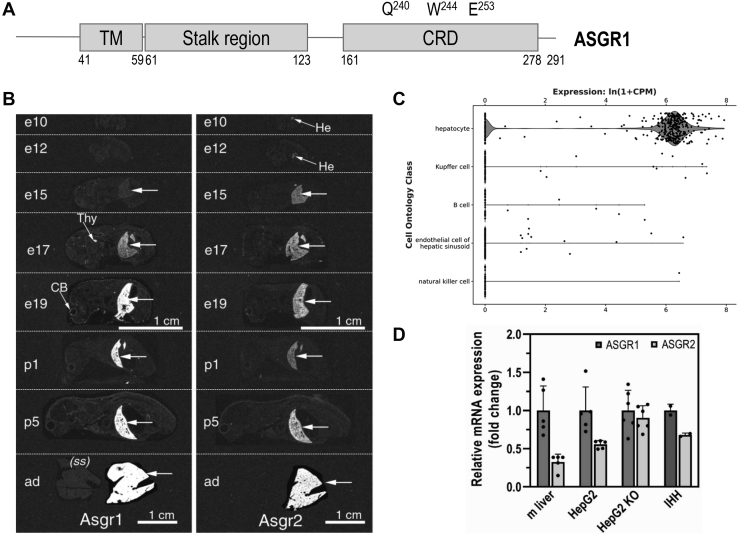
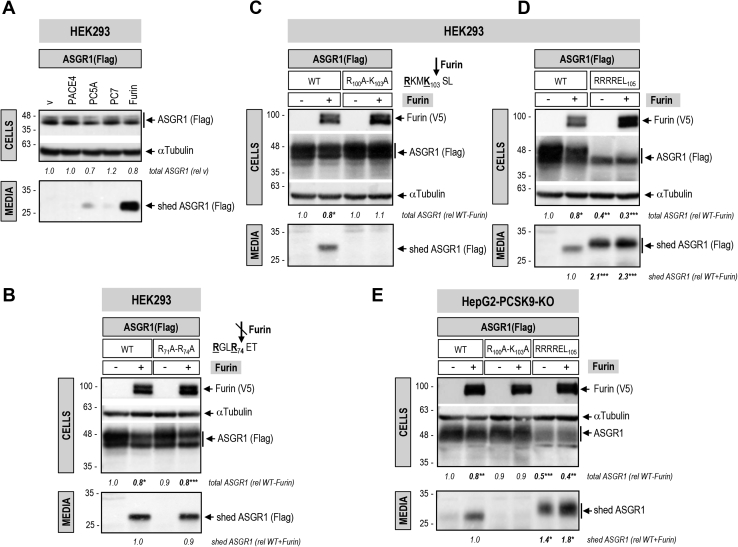
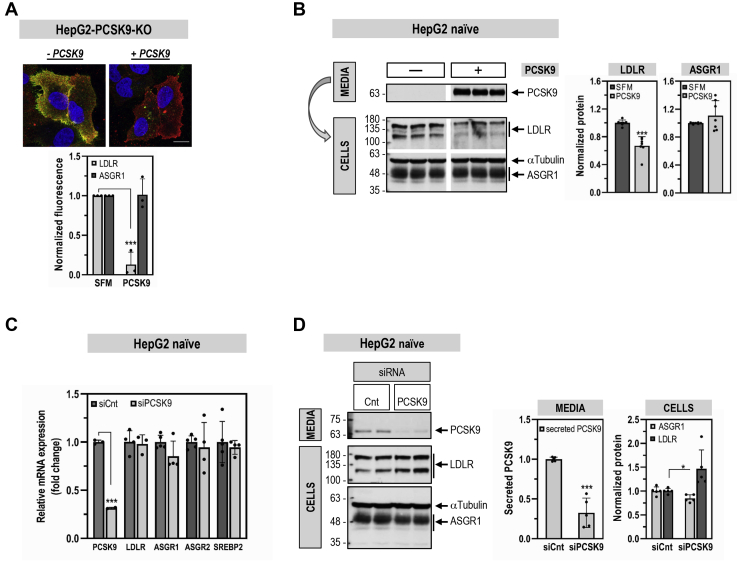
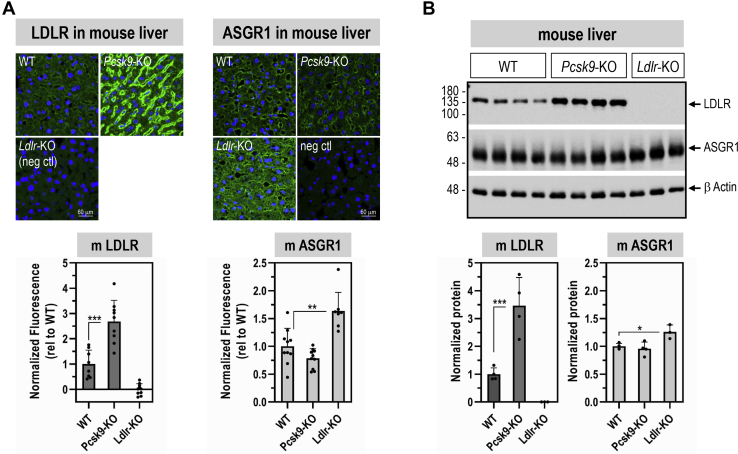
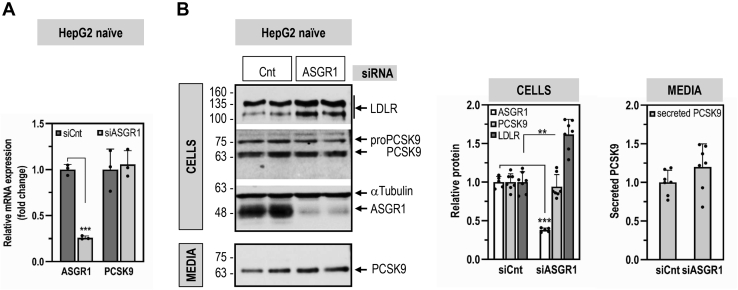
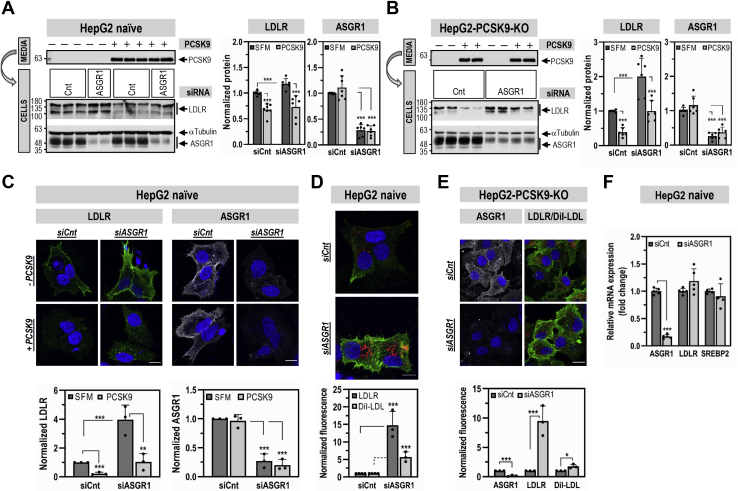
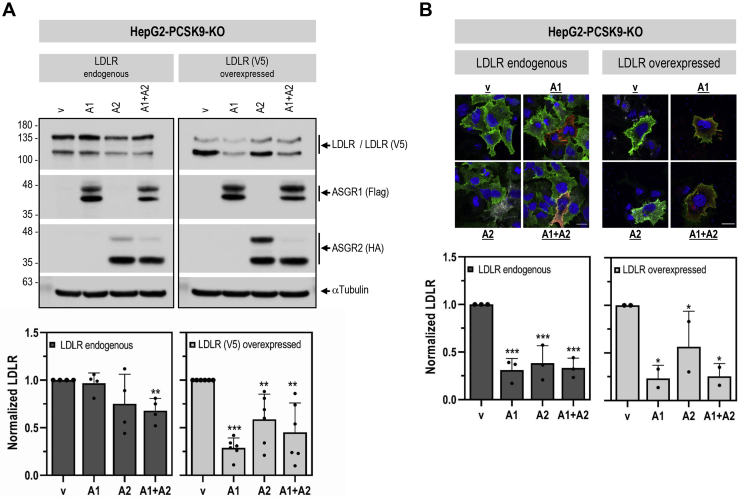
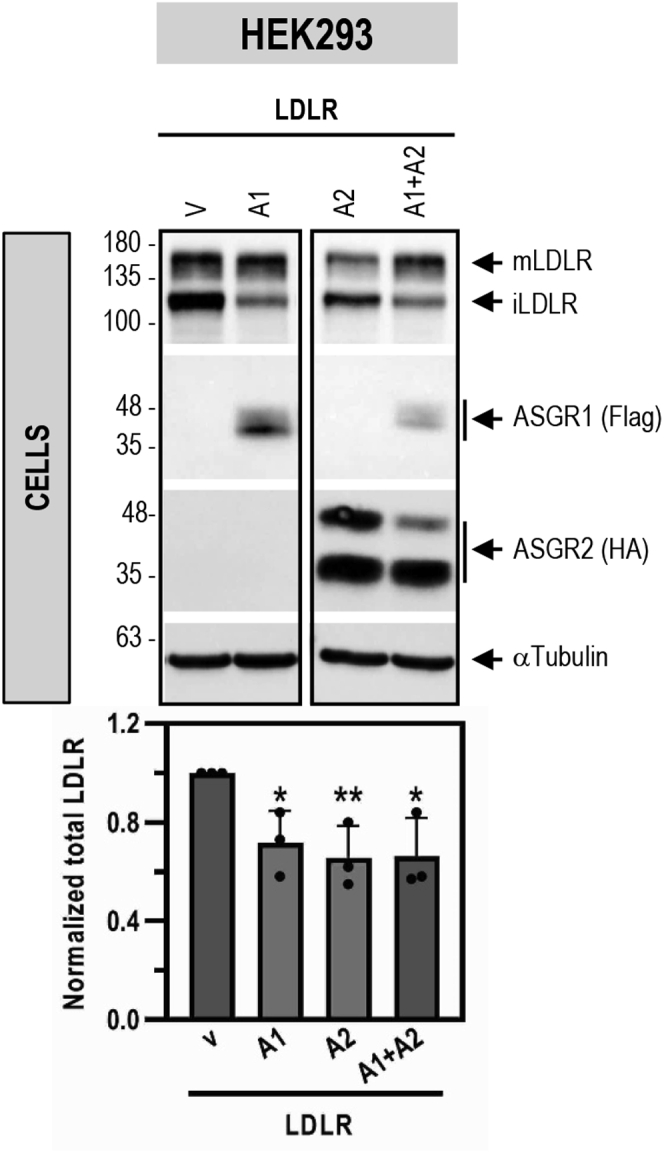
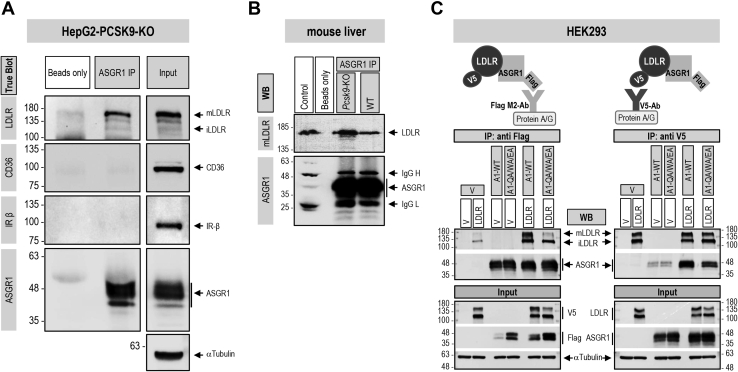
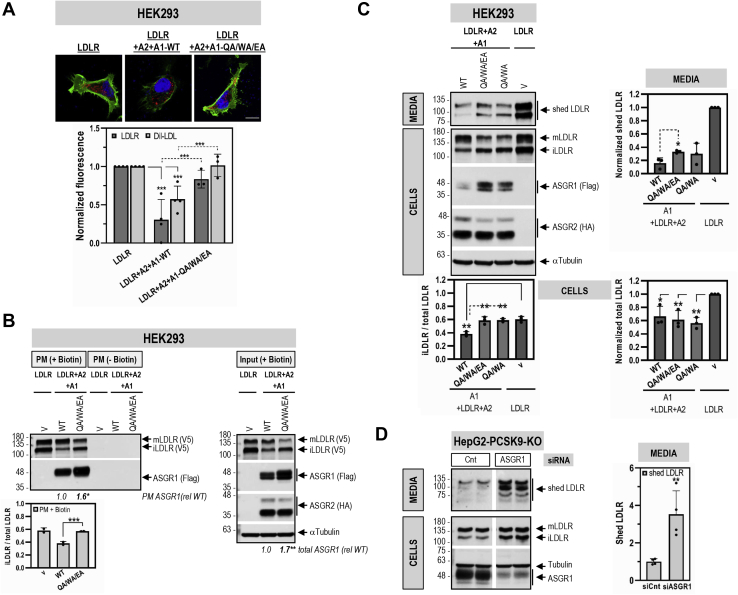
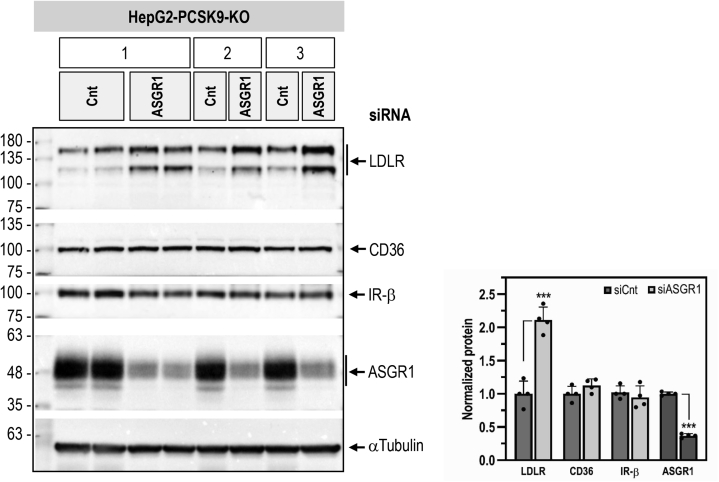
The hepatic carbohydrate-recognizing asialoglycoprotein receptor (ASGR1) mediates the endocytosis/lysosomal degradation of desialylated glycoproteins following binding to terminal galactose/N-acetylgalactosamine. Human heterozygote carriers of ASGR1 deletions exhibit ∼34% lower risk of coronary artery disease and ∼10% to 14% reduction of non-HDL cholesterol. Since the proprotein convertase PCSK9 is a major degrader of the low-density lipoprotein receptor (LDLR), we investigated the degradation and functionality of LDLR and/or PCSK9 by endogenous/overexpressed ASGR1 using Western blot and immunofluorescence in HepG2-naïve and HepG2-PCSK9-knockout cells. ASGR1, like PCSK9, targets LDLR, and both independently interact with/enhance the degradation of the receptor. This lack of cooperativity between PCSK9 and ASGR1 was confirmed in livers of wildtype (WT) and Pcsk9 mice. ASGR1 knockdown in HepG2-naïve cells significantly increased total (∼1.2-fold) and cell-surface (∼4-fold) LDLR protein. In HepG2-PCSK9-knockout cells, ASGR1 silencing led to ∼2-fold higher levels of LDLR protein and DiI (1,1'-dioctadecyl-3,3,3',3'-tetramethylindocarbocyanine perchlorate)-LDL uptake associated with ∼9-fold increased cell-surface LDLR. Overexpression of WT-ASGR1/2 primarily reduced levels of immature non-O-glycosylated LDLR (∼110 kDa), whereas the triple Ala-mutant of Gln240/Trp244/Glu253 (characterized by loss of carbohydrate binding) reduced expression of the mature form of LDLR (∼150 kDa), suggesting that ASGR1 binds the LDLR in both a sugar-dependent and -independent fashion. The protease furin cleaves ASGR1 at the RKMK↓ motif into a secreted form, likely resulting in a loss of function on LDLR. Altogether, we demonstrate that LDLR is the first example of a liver-receptor ligand of ASGR1. We conclude that silencing of ASGR1 and PCSK9 may lead to higher LDL uptake by hepatocytes, thereby providing a novel approach to further reduce LDL cholesterol levels.
肝脏糖识别性唾液酸糖蛋白受体 (ASGR1) 在与末端半乳糖/N-乙酰半乳糖胺结合后,介导去唾液酸化糖蛋白的内吞作用/溶酶体降解。ASGR1 缺失的杂合子携带者患冠状动脉疾病的风险降低约 34%,非高密度脂蛋白胆固醇降低 10%至 14%。由于前蛋白转化酶 PCSK9 是低密度脂蛋白受体 (LDLR) 的主要降解酶,我们使用 Western blot 和 HepG2-PCSK9 敲除细胞中的免疫荧光研究了内源性/过表达 ASGR1 对 LDLR 和/或 PCSK9 的降解和功能。ASGR1 与 PCSK9 一样,靶向 LDLR,并且两者独立地相互作用/增强受体的降解。这种 PCSK9 和 ASGR1 之间缺乏协同作用在野生型 (WT) 和 Pcsk9 小鼠的肝脏中得到了证实。在 HepG2-PCSK9 敲除细胞中,ASGR1 敲低显著增加了总(约 1.2 倍)和细胞表面(约 4 倍)LDLR 蛋白。在 HepG2-PCSK9 敲除细胞中,ASGR1 沉默导致 LDLR 蛋白水平升高约 2 倍,DiI(1,1'-二辛基-3,3,3',3'-四甲基吲哚羰花青高氯酸盐)-LDL 摄取增加约 9 倍,与细胞表面 LDLR 增加约 9 倍相关。WT-ASGR1/2 的过表达主要降低了不成熟的非 O-糖基化 LDLR(约 110 kDa)的水平,而 Gln240/Trp244/Glu253 的三 Ala 突变(特征是失去碳水化合物结合)降低了 LDLR 的成熟形式的表达(约 150 kDa),这表明 ASGR1 以糖依赖和非依赖的方式结合 LDLR。蛋白酶 furin 在 RKMK↓基序处切割 ASGR1 成分泌形式,可能导致 LDLR 功能丧失。总之,我们证明 LDLR 是 ASGR1 的第一个肝脏受体配体。我们得出结论,沉默 ASGR1 和 PCSK9 可能导致肝细胞摄取更多的 LDL,从而为进一步降低 LDL 胆固醇水平提供了一种新方法。