Department of Neonatology, Hunan Children's Hospital, No.86 Ziyuan Road, Changsha, 410007, China.
BMC Pediatr. 2021 Sep 20;21(1):414. doi: 10.1186/s12887-021-02875-x.
Lipoprotein lipase (LPL) deficiency is a monogenic lipid metabolism disorder biochemically characterized by hypertriglyceridemia (HTG) inherited in an autosomal recessive manner. Neonatal onset LPL deficiency is rare. The purpose of this study was to clarify the clinical features of neonatal LPL deficiency and to analyze the genetic characteristics of LPL gene.
In order to reach a definite molecular diagnose, metabolic diseases-related genes were sequenced through gene capture and next generation sequencing. Meanwhile, the clinical characteristics and follow-up results of the two newborns were collected and analyzed.
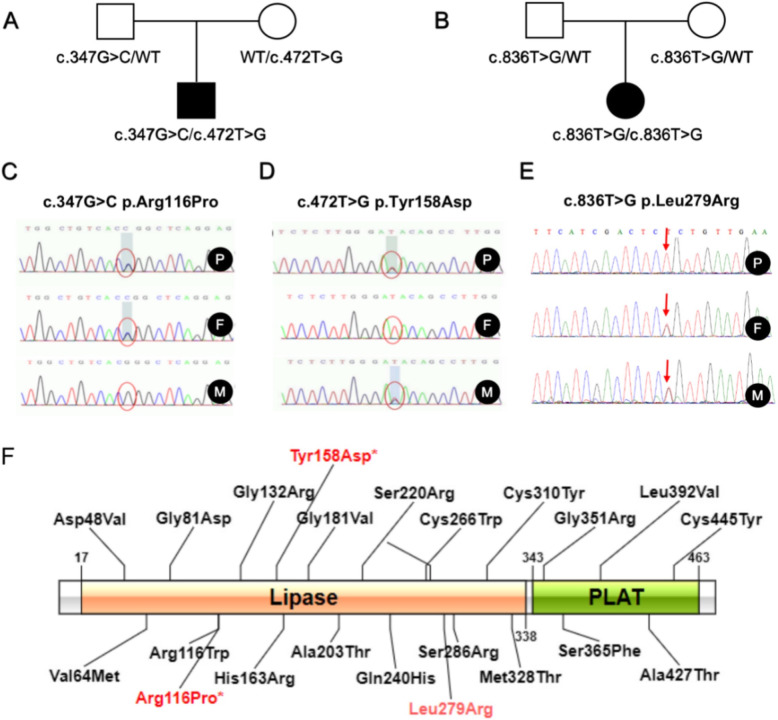
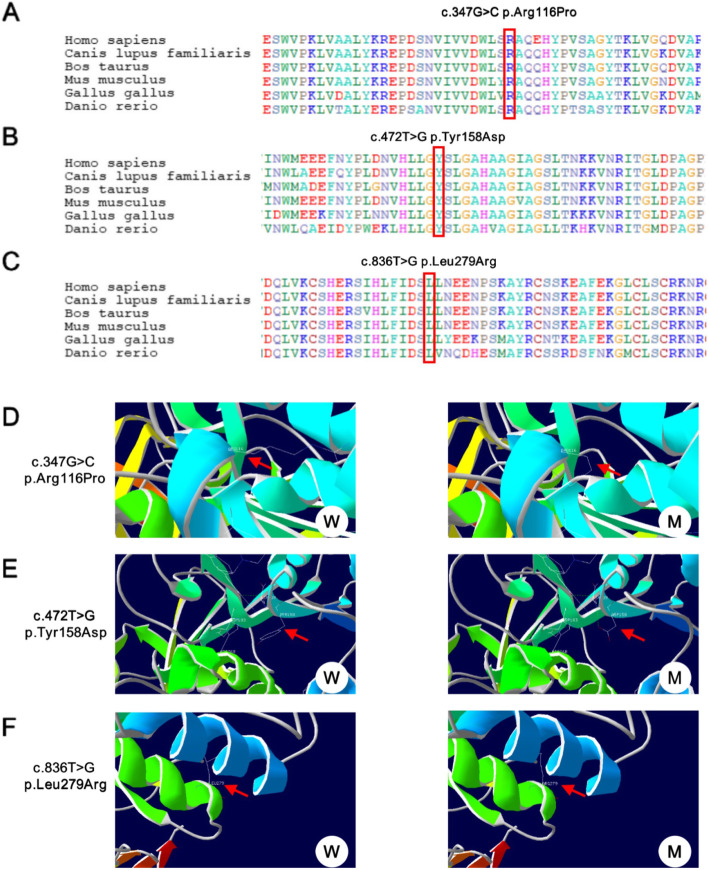
Three different mutations in the LPL gene were identified in the two newborns including a novel compound heterozygous mutation (c.347G > C and c.472 T > G) and a reported homozygous mutation (c.836 T > G) was identified. Interestingly, both the two neonatal onset LPL deficiency patients presented with suffered recurrent infection in the hyperlipidemia stage, which was not usually found in childhood or adulthood onset LPL deficiency patients.
The two novel mutaitons, c.347G > C and c.472 T > G, identified in this study were novel, which expanded the LPL gene mutation spectrum. In addition, suffered recurrent infection in the hyperlipidemia stage implied a certain correlation between immune deficiency and lipid metabolism abnormality. This observation further supplemented and expanded the clinical manifestations of LPL deficiency.
脂蛋白脂肪酶(LPL)缺乏症是一种单基因脂质代谢紊乱,其生化特征为常染色体隐性遗传的高甘油三酯血症(HTG)。新生儿期 LPL 缺乏症较为罕见。本研究旨在阐明新生儿 LPL 缺乏症的临床特征,并分析 LPL 基因的遗传特征。
为了达到明确的分子诊断,通过基因捕获和下一代测序对代谢疾病相关基因进行测序。同时,收集并分析了两名新生儿的临床特征和随访结果。
在两名新生儿中发现了 LPL 基因中的三个不同突变,包括一个新的复合杂合突变(c.347G>C 和 c.472T>G)和一个报道的纯合突变(c.836T>G)。有趣的是,两名新生儿 LPL 缺乏症患者在高脂血症期均出现反复感染,而在儿童期或成年期 LPL 缺乏症患者中通常不会出现这种情况。
本研究新发现的两个突变,c.347G>C 和 c.472T>G,均为新突变,扩大了 LPL 基因突变谱。此外,高脂血症期反复感染提示免疫缺陷与脂质代谢异常之间存在一定相关性。这一观察结果进一步补充和扩展了 LPL 缺乏症的临床表现。