Department of Medicine (Oncology), Albert Einstein College of Medicine-Montefiore Medical Center, Bronx, NY 10461.
InBios International Inc., Seattle, WA 98109.
Proc Natl Acad Sci U S A. 2021 Sep 28;118(39). doi: 10.1073/pnas.2106947118.
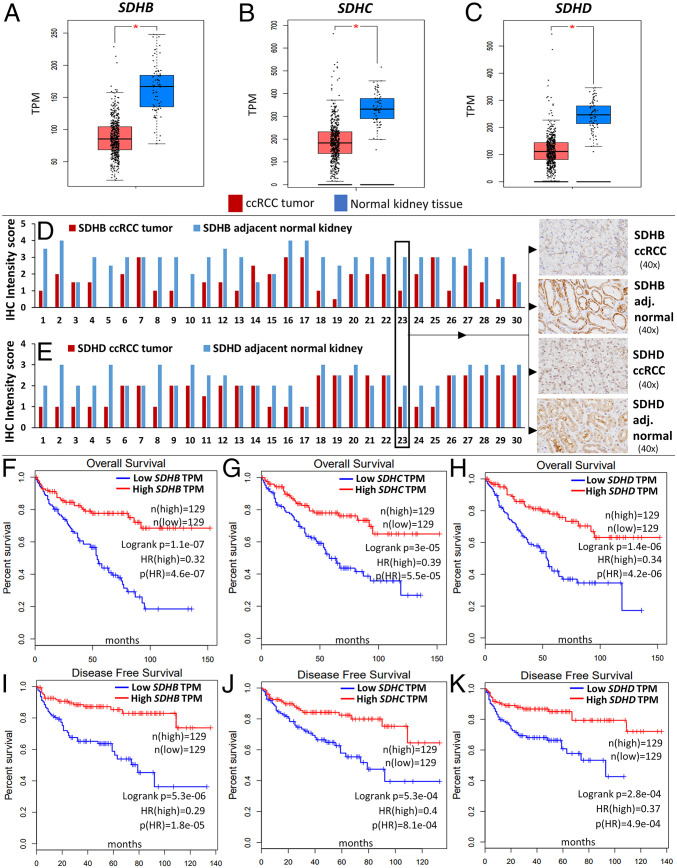
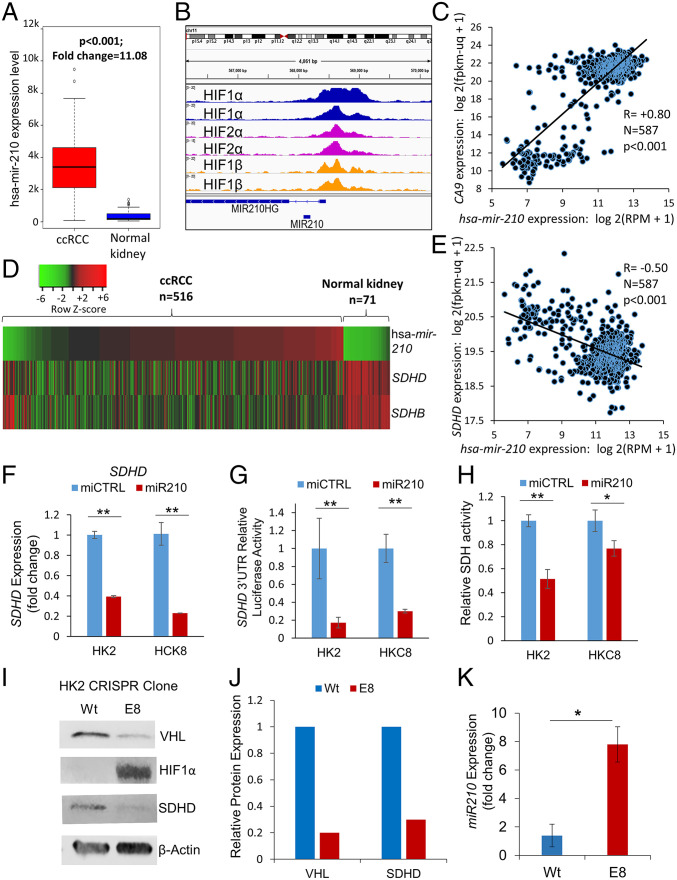
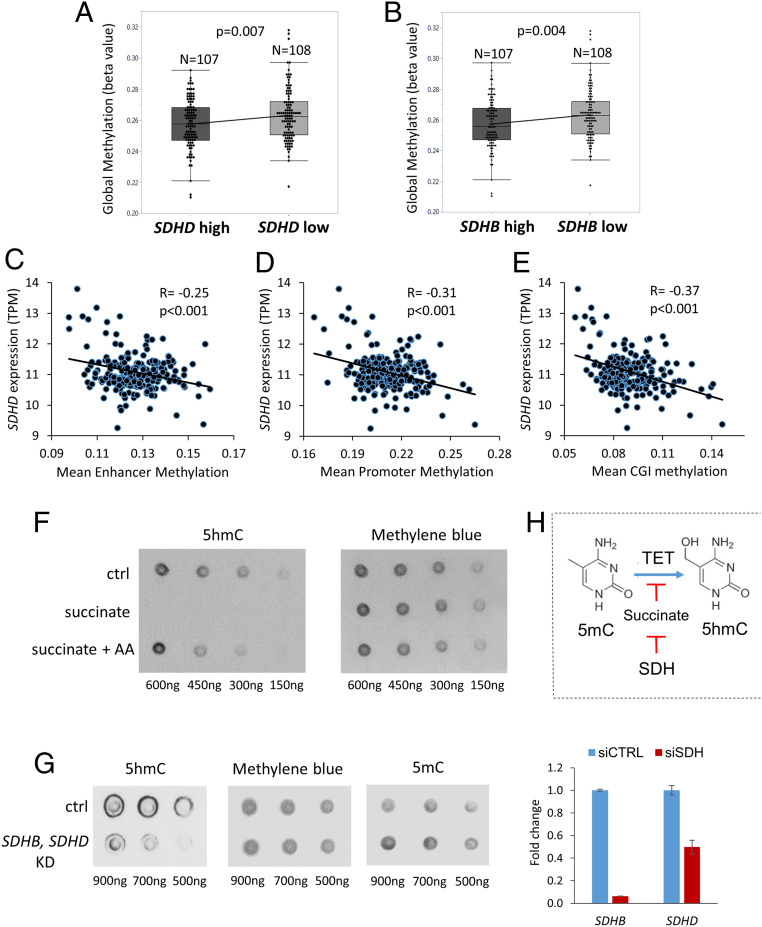
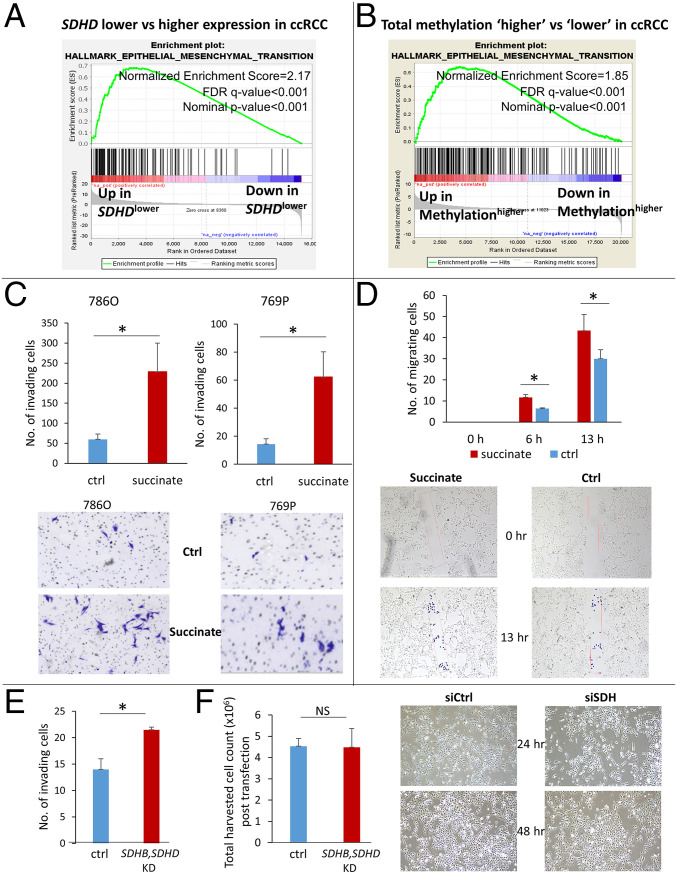
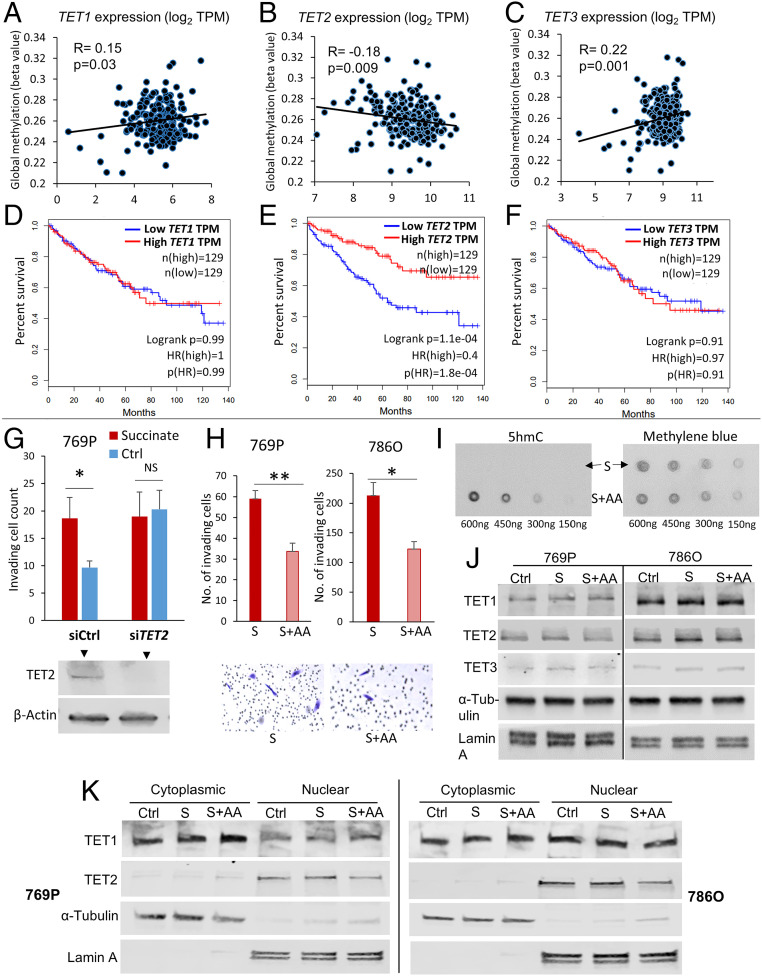
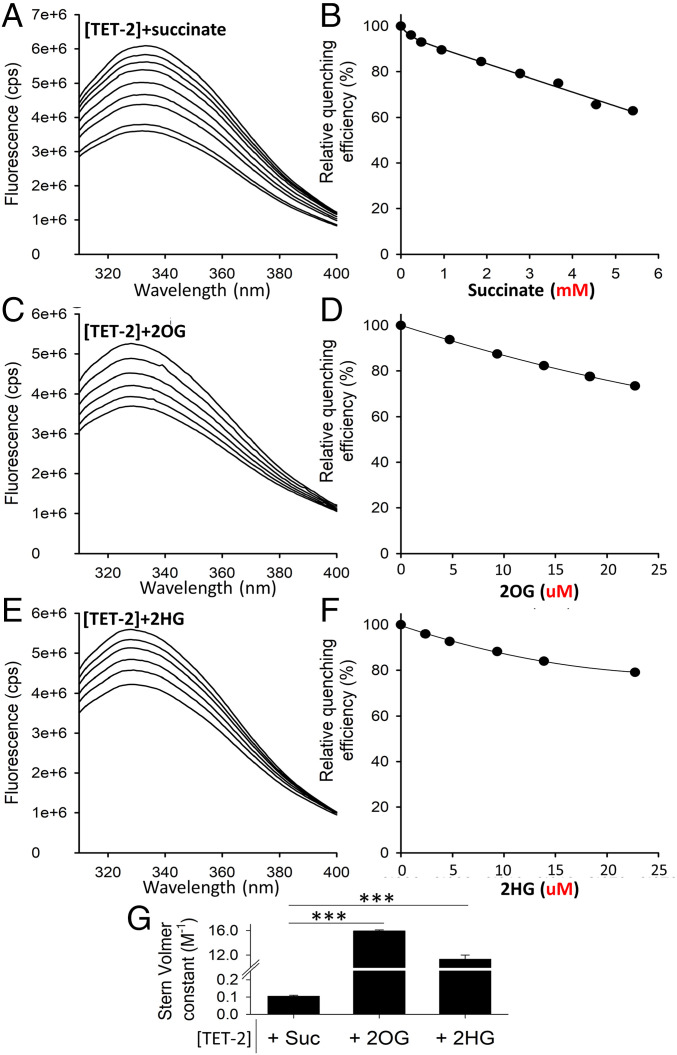
Reduced succinate dehydrogenase (SDH) activity resulting in adverse succinate accumulation was previously considered relevant only in 0.05 to 0.5% of kidney cancers associated with germline SDH mutations. Here, we sought to examine a broader role for SDH loss in kidney cancer pathogenesis/progression. We report that underexpression of SDH subunits resulting in accumulation of oncogenic succinate is a common feature in clear cell renal cell carcinoma (ccRCC) (∼80% of all kidney cancers), with a marked adverse impact on survival in ccRCC patients ( = 516). We show that SDH down-regulation is a critical brake in the TCA cycle during ccRCC pathogenesis and progression. In exploring mechanisms of SDH down-regulation in ccRCC, we report that Von Hippel-Lindau loss-induced hypoxia-inducible factor-dependent up-regulation of miR-210 causes direct inhibition of the transcript. Moreover, shallow deletion of occurs in ∼20% of ccRCC. We then demonstrate that SDH loss-induced succinate accumulation contributes to adverse loss of 5-hydroxymethylcytosine, gain of 5-methylcytosine, and enhanced invasiveness in ccRCC via inhibition of ten-eleven translocation (TET)-2 activity. Intriguingly, binding affinity between the catalytic domain of recombinant TET-2 and succinate was found to be very low, suggesting that the mechanism of succinate-induced attenuation of TET-2 activity is likely via product inhibition rather than competitive inhibition. Finally, exogenous ascorbic acid, a TET-activating demethylating agent, led to reversal of the above oncogenic effects of succinate in ccRCC cells. Collectively, our study demonstrates that functional SDH deficiency is a common adverse feature of ccRCC and not just limited to the kidney cancers associated with germline SDH mutations.
琥珀酸脱氢酶(SDH)活性降低导致琥珀酸积累不良,以前被认为仅与 0.05%至 0.5%与种系 SDH 突变相关的肾癌有关。在这里,我们试图研究 SDH 缺失在肾癌发病机制/进展中的更广泛作用。我们报告说,SDH 亚基表达降低导致致癌琥珀酸积累是透明细胞肾细胞癌(ccRCC)(所有肾癌的约 80%)的共同特征,对 ccRCC 患者的生存有明显的不利影响(=516)。我们表明,SDH 下调是 ccRCC 发病和进展过程中三羧酸循环的关键制动。在探索 ccRCC 中 SDH 下调的机制时,我们报告说,von Hippel-Lindau 缺失诱导的缺氧诱导因子依赖性 miR-210 上调导致对转录物的直接抑制。此外,在约 20%的 ccRCC 中发生了 浅缺失。然后,我们证明 SDH 缺失诱导的琥珀酸积累通过抑制 ten-eleven 易位(TET)-2 活性导致 ccRCC 中的 5-羟甲基胞嘧啶丢失、5-甲基胞嘧啶获得和侵袭性增强。有趣的是,发现重组 TET-2 的催化结构域与琥珀酸之间的结合亲和力非常低,这表明琥珀酸诱导的 TET-2 活性减弱的机制可能是通过产物抑制而不是竞争性抑制。最后,外源性抗坏血酸,一种 TET 激活的去甲基化剂,导致 ccRCC 细胞中琥珀酸的上述致癌作用逆转。总之,我们的研究表明,功能性 SDH 缺乏是 ccRCC 的常见不良特征,而不仅仅限于与种系 SDH 突变相关的肾癌。