Emory Vaccine Center, Department of Microbiology and Immunology, Emory University School of Medicine, Atlanta, GA 30322, USA.
Pharmaceutical Product Development, Wilmington, NC 28401, USA.
Viruses. 2021 Aug 27;13(9):1707. doi: 10.3390/v13091707.
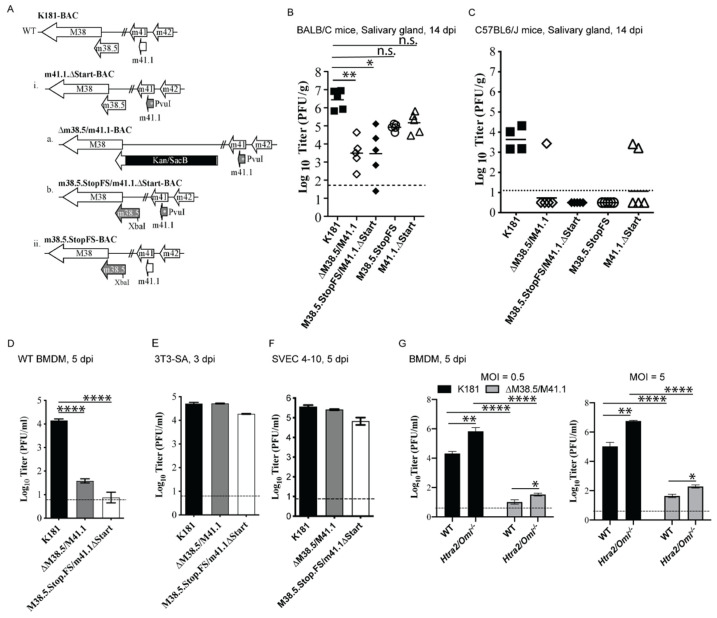
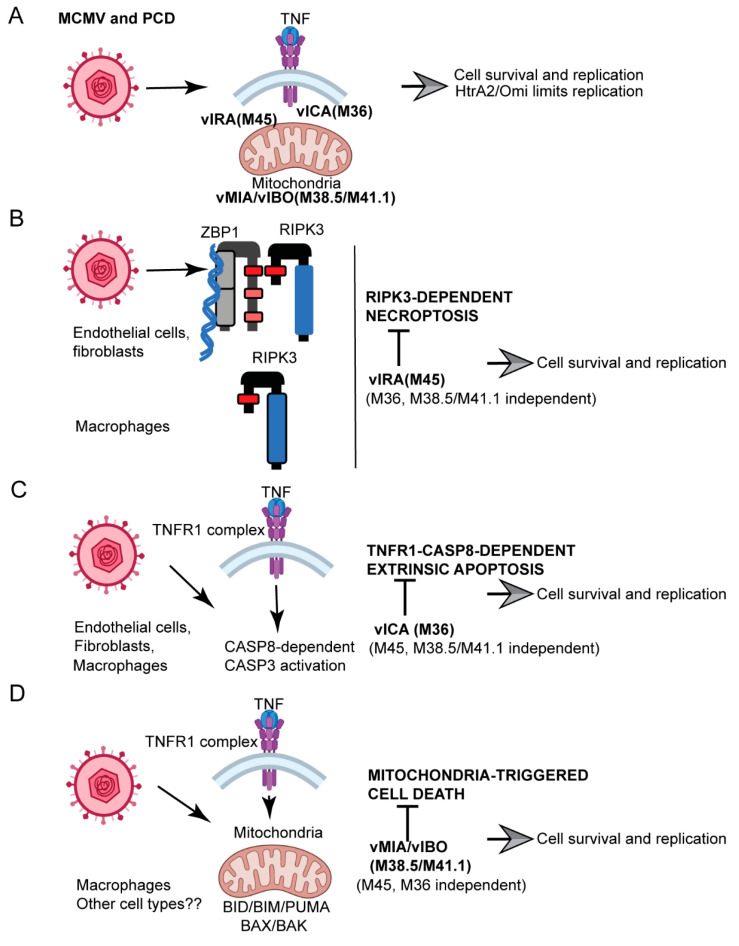
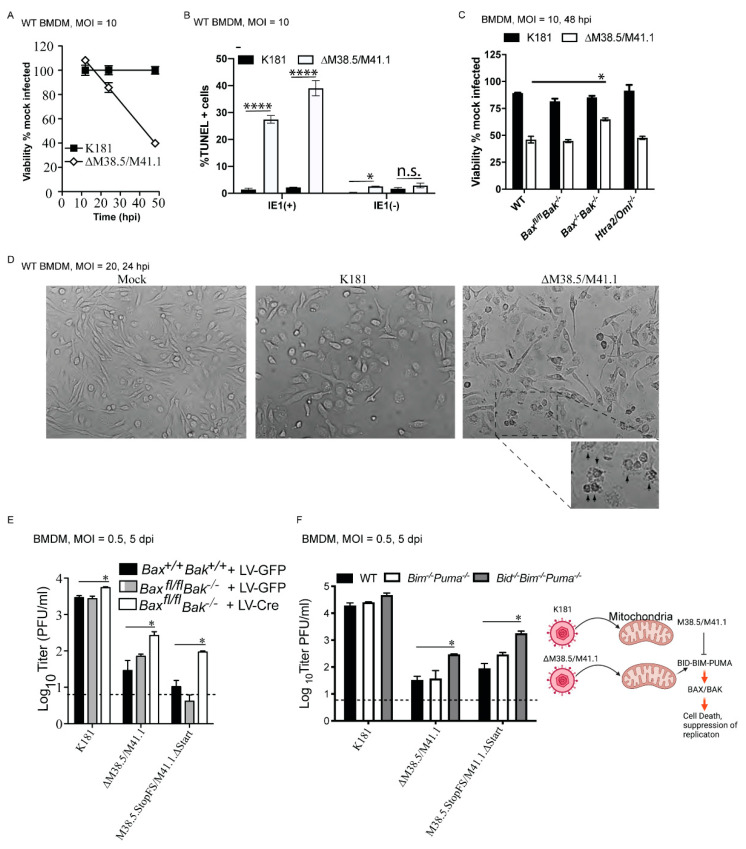
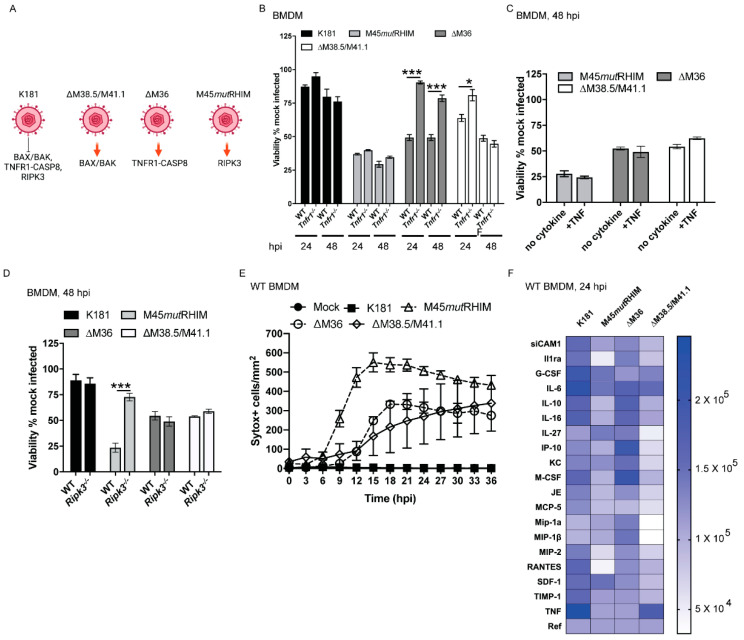
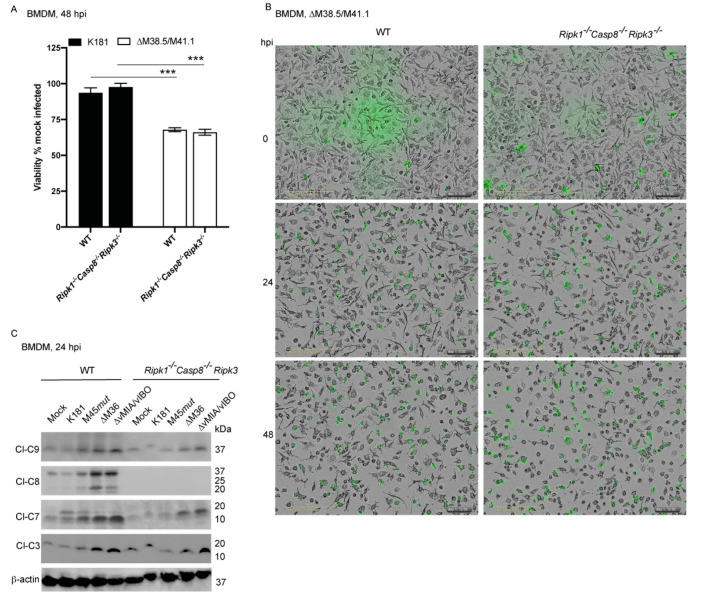
Programmed cell death pathways eliminate infected cells and regulate infection-associated inflammation during pathogen invasion. Cytomegaloviruses encode several distinct suppressors that block intrinsic apoptosis, extrinsic apoptosis, and necroptosis, pathways that impact pathogenesis of this ubiquitous herpesvirus. Here, we expanded the understanding of three cell autonomous suppression mechanisms on which murine cytomegalovirus relies: (i) M38.5-encoded viral mitochondrial inhibitor of apoptosis (vMIA), a BAX suppressor that functions in concert with M41.1-encoded viral inhibitor of BAK oligomerization (vIBO), (ii) M36-encoded viral inhibitor of caspase-8 activation (vICA), and (iii) M45-encoded viral inhibitor of RIP/RHIM activation (vIRA). Following infection of bone marrow-derived macrophages, the virus initially deflected receptor-interacting protein kinase (RIPK)3-dependent necroptosis, the most potent of the three cell death pathways. This process remained independent of caspase-8, although suppression of this apoptotic protease enhances necroptosis in most cell types. Second, the virus deflected TNF-mediated extrinsic apoptosis, a pathway dependent on autocrine TNF production by macrophages that proceeds independently of mitochondrial death machinery or RIPK3. Third, cytomegalovirus deflected BCL-2 family protein-dependent mitochondrial cell death through combined TNF-dependent and -independent signaling even in the absence of RIPK1, RIPK3, and caspase-8. Furthermore, each of these cell death pathways dictated a distinct pattern of cytokine and chemokine activation. Therefore, cytomegalovirus employs sequential, non-redundant suppression strategies to specifically modulate the timing and execution of necroptosis, extrinsic apoptosis, and intrinsic apoptosis within infected cells to orchestrate virus control and infection-dependent inflammation. Virus-encoded death suppressors together hold control over an intricate network that upends host defense and supports pathogenesis in the intact mammalian host.
程序性细胞死亡途径可清除受感染的细胞,并在病原体入侵时调节感染相关的炎症。巨细胞病毒编码了几种不同的抑制剂,可阻断内在凋亡、外在凋亡和坏死性凋亡,这些途径会影响这种普遍存在的疱疹病毒的发病机制。在这里,我们扩展了对三种依赖于鼠巨细胞病毒的细胞自主抑制机制的理解:(i)M38.5 编码的病毒线粒体凋亡抑制剂(vMIA),一种 BAX 抑制剂,与 M41.1 编码的病毒 BAK 寡聚化抑制剂(vIBO)协同作用;(ii)M36 编码的病毒半胱天冬酶-8 激活抑制剂(vICA);和(iii)M45 编码的病毒 RIP/RHIM 激活抑制剂(vIRA)。在骨髓来源的巨噬细胞感染后,病毒最初回避受体相互作用蛋白激酶(RIPK)3 依赖性坏死性凋亡,这是三种细胞死亡途径中最有效的一种。这个过程仍然不依赖于半胱天冬酶-8,尽管抑制这种凋亡蛋白酶会增强大多数细胞类型的坏死性凋亡。其次,病毒回避 TNF 介导的外在凋亡,这是一种依赖于巨噬细胞自分泌 TNF 产生的途径,该途径独立于线粒体死亡机制或 RIPK3。第三,巨细胞病毒通过联合 TNF 依赖和非依赖信号转导回避 BCL-2 家族蛋白依赖性线粒体细胞死亡,即使在缺乏 RIPK1、RIPK3 和半胱天冬酶-8 的情况下也是如此。此外,这些细胞死亡途径中的每一种都决定了细胞因子和趋化因子激活的独特模式。因此,巨细胞病毒采用顺序的、非冗余的抑制策略来特异性调节受感染细胞中坏死性凋亡、外在凋亡和内在凋亡的时间和执行,以协调病毒控制和感染依赖性炎症。病毒编码的死亡抑制剂共同控制着一个复杂的网络,颠覆了宿主防御,并在完整的哺乳动物宿主中支持发病机制。